



Иматиниб – противоопухолевый препарат нового класса таргетных цитостатиков, активно применяемый при лечении меланомы и миелоидного лейкоза. Иматиниб впервые был разработан швейцарской компанией Novartis и реализован под торговым наименованием Гливек®[4]. Для государственной регистрации дженерика, в частности и препарата на основе иматиниба, в Российской Федерации необходимо подтверждение его эффективности и безопасности оригинальному препарату, которое устанавливается путем исследования биоэквивалентности [7]. Исследование биоэквивалентности является сложным и многогранным, включающим в себя медицинскую составляющую, аналитическое сопровождение, расчет важнейших фармакокинетических параметров и статистическую обработку результатов.
Целью настоящей работы являлось исследование биоэквивалентности нового воспроизведенного лекарственного препарата IMB (кодовое название) и оригинального лекарственного препарата Гливек® (Novartis Pharma Stein AG, Швейцария) после однократного перорального приема здоровыми добровольцами.
Материалы и методы исследования
Объектами исследования являлись тест-препарат (Т) – IMB, капсулы 100 мг (Россия) и референс-препарат (R) – Гливек®, капсулы 100 мг (Novartis Pharma Stein AG, Швейцария).
Исследование препаратов выполнялось в соответствии с протоколом, Методическими указаниями Минздравсоцразвития «Проведение качественных исследований биоэквивалентности лекарственных средств» (Россия, 2008 г.) [5] и регламентировалось документами действующего законодательства РФ [1, 8]. В качестве добровольцев были привлечены 24 мужчины в возрасте от 18 до 45 лет, отобранные согласно критериям включения и невключения.
Дизайн исследования
Проведенное исследование являлось рандомизированным, открытым, перекрестным, сравнительным, с двумя периодами однократного введения препарата добровольцам и периодом отмывки продолжительностью 14 суток.
В ходе исследования осуществляли динамическое наблюдение за добровольцами (измерение артериального давления, частоты сердечных сокращений, частоты дыхания). До приема препарата, утром, натощак путем внутривенного катетера отбирали пробу крови для оценки исходного статуса. Через 30 мин после окончания приема пищи добровольцы осуществли пероральный прием 4 капсул (400 мг) одного из препаратов, запивая 200 мл кипяченой воды комнатной температуры. В дальнейшем отбор проб крови проводили через 30 мин и 1, 1,5, 2, 3, 4, 6, 8, 10, 12, 24, 48, 72 ч. Отбор крови через 24, 48 и 72 ч проводили путем венопункции. Второй период исследования проводили через 14 дней по идентичной схеме.
Аналитическая методика
Количественное определение иматиниба в плазме крови добровольцев проводили методом ВЭЖХ с УФ-детектированием на приборе Милихром А-02 (ЗАО «ЭкоНова», Новосибирск, Россия) по разработанной и валидированной аналитической методике [2–3].
В качестве пробоподготовки для извлечения иматиниба из биологической матрицы использовали метод жидкостно-жидкостной экстракции ацетонитрилом по принципу QuEChERS (Quick, Easy, Cheap, Effective, Rugged, Safe). В центрифужную микропробирку вместимостью 2 мл помещали 500 мкл плазмы крови, содержащей иматиниб, прибавляли 1 мл ацетонитрила и тщательно перемешивали на встряхивателе Elmi V-3 при скорости 4000 об/мин в течение 10 мин. Затем к раствору прибавляли 0,1–0,2 г натрия хлорида и перемешивали в течение 5 мин. Полученный раствор центрифугировали в центрифуге Heraeus Biofuge pico (Германия) при 13000 об/мин в течение 10 мин. Водную фазу отбрасывали, а органическую упаривали на вакуумном концентраторе UniEquip Univapo 100 ECH (Германия) при температуре 45 °С в течение 80 мин. К сухому остатку прибавляли 500 мкл подвижной фазы А, перемешивали 5 мин и центрифугировали при 13000 об/мин в течение 5 мин. Надосадочную жидкость использовали для хроматографического анализа.
Разделение проводили на колонке размером 75×2 мм, заполненной сорбентом ProntoSIL C18, 5 мкм, при температуре 35 °С и скорости потока 0,1 мл/мин. Состав подвижной фазы: А – 0,5 % раствор калия фосфорнокислого однозамещенного – метанол – триэтиламин 74:25:1 с рН равным 3,3, доведенным ортофосфорной кислотой; Б – метанол с рН равным 3,3, доведенным ортофосфорной кислотой. Объем вводимой пробы – 20 мкл, длина волны УФ-детектирования – 260 нм. Условия градиентного режима элюирования представлены в табл. 1.
Таблица 1
Условия градиентного режима элюирования
|
Ступень градиента |
Время, мин |
Доля элюента А, % |
Доля элюента Б, % |
|
Кондиционирование |
15 |
95 |
5 |
|
0 |
0 |
95 |
5 |
|
1 |
10 |
30 |
70 |
|
2 |
20 |
30 |
70 |
Нижний предел количественного определения иматиниба в плазме крови, согласно данной методике, составляет 0,042 мкг/мл.
Для подтверждения пригодности проведена валидация разработанной биоаналитической методики на основании «Руководства по экспертизе лекарственных средств. Том 1» (Россия, 2013) и согласно требованиям руководств по валидации биоаналитических методик «Guidance for Industry: Bioanalytical method validation» (FDA, США, 2001) и «Guideline on validation of bioanalytical methods» (EMA, Англия, 2009) по следующим характеристикам: селективность, линейность, правильность и прецизионность, перенос, стабильность.
Фармакокинетический анализ
Индивидуальные профили изменения концентраций (С) иматиниба в плазме крови добровольца во времени (t), зарегистрированные после приема препаратов IMB и Гливек®, характеризовали максимальной концентрацией лекарственного вещества (Cmax) и временем достижения максимальной концентрации (Tmax), а также площадью под кривой «концентрация – время» в пределах от нуля до момента последнего отбора пробы крови (t = 72 ч), рассчитанной методом трапеций (AUC0-72). Сопоставление значений AUC0-72 c AUC∞,в случае AUC0-72 0,8 AUC≥, свидетельствовало о том, что выбранный регламент фармакокинетического исследования обеспечивает необходимую надежность оценки биодоступности иматиниба. В качестве оценки относительной степени всасывания (f) лекарственного вещества использовали отношение AUC0-,T/AUC0-,R, а относительной биодоступности (f″) исследуемого препарата по отношению к препарату сравнения – 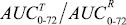 . Относительную степень всасывания (f″) исследуемого препарата по отношению к препарату сравнения определяли отношением
. Относительную степень всасывания (f″) исследуемого препарата по отношению к препарату сравнения определяли отношением 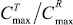 .
.
Статистический анализ
Статистическая обработка была выполнена в соответствии с Методическими указаниями «Оценка биоэквивалентности лекарственных средств» (Москва, 2008) с помощью статистического пакета Microsoft Excel 2013. Гипотеза о биоэквивалентности формулировалась в терминах отношения сравниваемых параметров:
– нулевая гипотеза об отсутствии эквивалентности H0: mT/mR < Q1 или mT/mR > Q2;
– соответствующая альтернативная гипотеза о наличии биоэквивалентности HA: Q1 mT/mR Q2;
где Q1 и Q2 – нижняя и верхняя принятые допустимые границы биоэквивалентности, а mT и mR – генеральные средние показатели сравнения для испытуемого лекарственного средства и препарата сравнения соответственно. Предполагалось, что AUC0-t, Cmax и Cmax/AUC0-t имеют логнормальное распределение. После проведения логарифмического преобразования эти показатели анализировались с помощью дисперсионного анализа (ANOVA). При этом учитывались следующие факторы, вносящие вклад в наблюдаемую вариацию данных:
– различия между испытуемыми (межиндивидуальные различия);
– различия между лекарственными средствами;
– последовательность приема лекарственных средств;
– периоды полувыведения.
Препараты признавались биоэквивалентными, если 90 % доверительные интервалы для значений площади под фармакокинетической кривой AUC0-t, максимальной концентрации Cmax и отношения Cmax/AUC0-t находились в следующих диапазонах:
80 < AUC0-t < 125,
75 < Cmax < 133,
75 < Cmax/AUC0-t < 133.
Для всех статистических процедур в данном исследовании вероятность совершения ошибки первого рода a была принята равной 0,05.
Статистическим критерием прекращения исследования биоэквивалентности являлось включение запланированного количества добровольцев – обеспечение требуемого уровня статистической мощности.
Результаты исследования и их обсуждение
С помощью разработанной и валидированной методики количественного определения иматиниба в плазме крови методом ВЭЖХ [2–3] проведен анализ образцов плазмы крови добровольцев после приема тестируемого и референтного препаратов. Построенные по результатам анализа образцов фармакокинетические кривые иматиниба в плазме крови добровольцев после однократного перорального приема препаратов IMB и Гливек в дозе 400 мг представлены на рисунке.
Из рисунка видно, что усредненные фармакокинетические профили иматиниба после однократного приема 4 капсул препаратов IMB и Гливек® практически совпадают.
На основании полученных результатов анализа рассчитаны фармакокинетические параметры, представленные в табл. 2 и 3.
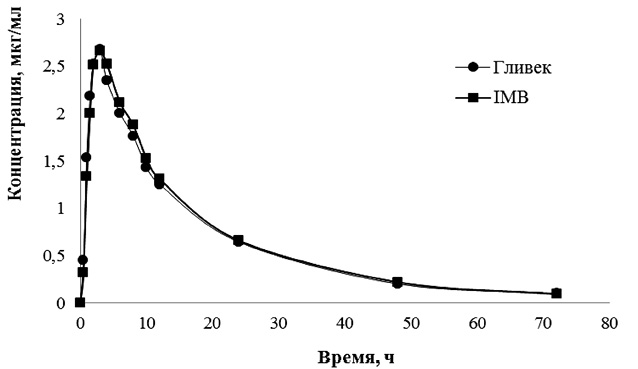
Динамика усредненных концентраций иматиниба (нг/мл) после приема тестируемого и референтного препаратов в линейных координатах
Таблица 2
Фармакокинетические параметры иматиниба после приема 4 капсул исследуемого препарата IMB
|
Параметр |
Cmax, мкг/мл |
Tmax, ч |
AUC0-72 |
AUC0-0-∞ |
Kel, ч–1 |
T1/2 |
AUMC |
MRT |
AUC0-72/AUC0-0-∞ |
Cmax/AUC0-72 |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
|
Среднее арифметическое |
2,87 |
3,04 |
48,75 |
51,18 |
0,05 |
15,9 |
1089,6 |
22,3 |
95,32 |
0,06 |
|
Среднее геометрическое |
2,75 |
2,91 |
47,10 |
49,43 |
0,04 |
13,9 |
1025,6 |
21,8 |
95,28 |
0,06 |
|
Стандартное отклонение |
0,87 |
0,95 |
13,40 |
14,03 |
0,06 |
5,54 |
409,18 |
5,35 |
3,06 |
0,01 |
|
Коэффициент вариации, % |
30,4 |
31,4 |
27,49 |
27,42 |
104,4 |
34,9 |
37,55 |
24,0 |
3,21 |
15,12 |
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
|
Медиана |
2,74 |
3,0 |
46,21 |
48,20 |
0,04 |
16,4 |
963,81 |
21,0 |
96,01 |
0,06 |
|
Минимальное |
1,53 |
2,0 |
28,11 |
28,49 |
0,03 |
1,21 |
433,19 |
15,4 |
86,03 |
0,04 |
|
Максимальное |
4,54 |
6,0 |
78,10 |
82,69 |
0,31 |
26,8 |
2240,4 |
39,8 |
100,0 |
0,08 |
|
Первый квартиль |
2,25 |
2,0 |
42,17 |
43,86 |
0,04 |
14,5 |
824,0 |
18,3 |
93,49 |
0,05 |
|
Третий квартиль |
3,34 |
3,25 |
52,10 |
57,86 |
0,05 |
18,7 |
1242,9 |
24,9 |
96,95 |
0,07 |
|
Размах |
3,0 |
4,0 |
49,99 |
54,20 |
0,28 |
25,5 |
1807,2 |
24,4 |
13,97 |
0,03 |
Таблица 3
Фармакокинетические параметры иматиниба после приема 4 капсул исследуемого препарата Гливек®
|
Параметр |
Cmax, мкг/мл |
Tmax, ч |
AUC0-72 |
AUC0-0-∞ |
Kel, ч–1 |
T1/2 |
AUMC |
MRT |
AUC0-72/AUC0-0-∞ |
Cmax/AUC0-72 |
|
Среднее арифметическое |
2,79 |
2,63 |
47,12 |
50,29 |
0,04 |
18,1 |
1150,2 |
23,9 |
94,17 |
0,06 |
|
Среднее геометрическое |
2,69 |
2,49 |
45,60 |
48,49 |
0,04 |
17,3 |
1045,8 |
22,9 |
94,06 |
0,06 |
|
Стандартное отклонение |
0,76 |
0,81 |
12,29 |
14,06 |
0,01 |
5,59 |
604,64 |
8,50 |
4,50 |
0,01 |
|
Коэффициент вариации, % |
27,2 |
30,9 |
26,08 |
27,96 |
34,06 |
30,9 |
52,57 |
35,5 |
4,78 |
15,67 |
|
Медиана |
2,69 |
3,0 |
45,15 |
48,36 |
0,04 |
17,9 |
1061,2 |
21,8 |
94,73 |
0,06 |
|
Минимальное |
1,48 |
1,0 |
21,53 |
21,92 |
0,02 |
8,82 |
370,05 |
14,9 |
78,26 |
0,04 |
|
Максимальное |
4,33 |
4,0 |
80,99 |
92,53 |
0,08 |
34,2 |
2969,8 |
56,6 |
100,0 |
0,08 |
|
Первый квартиль |
2,19 |
2,0 |
39,51 |
42,21 |
0,03 |
14,8 |
833,93 |
19,6 |
92,50 |
0,05 |
|
Третий квартиль |
3,32 |
3,0 |
50,49 |
54,17 |
0,05 |
20,4 |
1214,4 |
25,8 |
96,74 |
0,07 |
|
Размах |
2,85 |
3,0 |
59,46 |
70,60 |
0,06 |
25,3 |
2599,8 |
41,7 |
21,74 |
0,04 |
Таблица 4
Относительная биодоступность и относительная степень всасывания иматиниба после приема 400 мг (4 капсул) исследуемого препарата IMB по сравнению с приемом препарата сравнения Гливек®
|
Параметр |
f, % |
f, % |
f, % |
|
Среднее арифметическое |
103,42 |
104,75 |
104,34 |
|
Среднее геометрическое |
101,95 |
103,28 |
102,20 |
|
Стандартное отклонение |
18,87 |
19,03 |
21,75 |
|
Коэффициент вариации, % |
18,25 |
18,17 |
20,85 |
|
Медиана |
100,59 |
100,27 |
102,73 |
|
Минимальное |
80,26 |
78,29 |
67,96 |
|
Максимальное |
157,40 |
157,40 |
147,08 |
|
Первый квартиль |
91,19 |
94,26 |
88,52 |
|
Третий квартиль |
107,62 |
109,48 |
116,40 |
|
Размах |
77,14 |
79,11 |
79,12 |
Таблица 5
Результаты дисперсионного анализа
|
Источник вариации |
SS |
DF |
MS |
F |
P |
|
ln AUC0-t |
|||||
|
Препарат |
0,012471 |
1 |
0,012471 |
1,125507 |
0,300244 |
|
Последовательность |
0,025728 |
1 |
0,025728 |
2,321972 |
0,141805 |
|
Испытуемые |
2,924574 |
23 |
0,127155 |
11,475734 |
1,30·10-7 |
|
Период |
0,055402 |
1 |
0,055402 |
5,000056 |
0,035812 |
|
Остаточная вариация |
0,243768 |
22 |
0,011080 |
– |
– |
|
Общая вариация |
3,261944 |
47 |
– |
– |
– |
|
ln Cmax |
|||||
|
Препарат |
0,005685 |
1 |
0,005685 |
0,298626 |
0,590245 |
|
Последовательность |
0,013264 |
1 |
0,013264 |
0,696724 |
0,412859 |
|
Испытуемые |
3,382733 |
23 |
0,147075 |
7,725251 |
0,000005 |
|
Период |
0,067825 |
1 |
0,067825 |
3,562554 |
0,072360 |
|
Остаточная вариация |
0,418842 |
22 |
0,019038 |
– |
– |
|
Общая вариация |
3,888350 |
47 |
– |
– |
– |
|
ln Cmax/AUC0-t |
|||||
|
Препарат |
0,001316 |
1 |
0,001316 |
0,159529 |
0,693442 |
|
Последовательность |
0,002046 |
1 |
0,002046 |
0,248038 |
0,623402 |
|
Период |
0,925996 |
23 |
0,040261 |
4,881586 |
0,000210 |
|
Испытуемые |
0,000628 |
1 |
0,000628 |
0,076113 |
0,785210 |
|
Остаточная вариация |
0,181444 |
22 |
0,008247 |
– |
– |
|
Общая вариация |
1,111429 |
47 |
– |
– |
– |
Из табл. 2 и 3 следует, что значения отношений AUC0-t/AUC0-0-∞ превышают 80 %, что говорит о достаточной длительности наблюдения, поэтому дальнейшие расчеты проводили с использованием показателя биодоступности AUC0-t, результаты которых представлены в табл. 4.
Относительная степень всасывания иматиниба для препарата IMB и величина отношения максимальных концентраций после приема испытуемого препарата и препарата сравнения статистически отличались незначительно.
Для проверки гипотез о статистической значимости вклада каждого из выбранных факторов (различия между препаратами, между добровольцами, последовательность приема препаратов) в наблюдаемую вариабельность данных применялся дисперсионный анализ ANOVA, результаты которого представлены в табл. 5.
Достоверный вклад в наблюдавшуюся вариабельность сравниваемых параметров вносила разница между добровольцами для параметров Cmax и AUC0-t. Вклад периода приема препаратов для параметров Cmax/AUC0-t и AUC0-t также являлся достоверным. Вклад различий между препаратами и последовательностью их приема был недостоверным для всех трех параметров. Оценка биоэквивалентности по этим параметрам позволяет сделать вывод о биоэквивалентности изученных препаратов, как по полноте, так и по скорости всасывания.
Заключение
Таким образом, проведенное исследование позволяет сделать вывод о биоэквивалентности препаратов IMB, капсулы 100 мг (Россия) и Гливек®, капсулы 100 мг (Novartis Pharma Stein AG, Швейцария) после однократного перорального приема здоровыми добровольцами и предположить сопоставимость клинической эффективности и безопасности этих препаратов у пациентов. Проблему обеспечения иматинибом больных в нашей стране позволит решить появление на российском рынке воспроизведенного лекарственного препарата на его основе.
Следует отметить, что полученные нами данные согласуются с результатами работ, в которых также была изучена биоэквивалентность препаратов иматиниба различных производителей [6, 9].