


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
RESEARCH OF CONGENITAL FAULTS OF DEVELOPMENTAL AND HEREDITARY DISEASE IN WESTERN REGIONS OF AZERBAIJAN

Вся наследственная патология определяется грузом мутаций, возникающих и унаследованных из предыдущих поколений. Эффекты мутационного процесса для популяции человека выражаются в уровне заболеваемости врожденными пороками развития (ВПР) и наследственными заболеваниями (НЗ) [1]. Так, установлено, что около 10% населения имеют наследственные нарушения [2]. По данным ВОЗ 2,5% всех новорожденных - с пороками развития, 1,5% из них обусловлены действием неблагоприятных экзогенных факторов во время беременности, остальные имеют преимущественно генетическую природу. Около 40-50% перинатальной смертности и инвалидности с детства также обусловлены наследственными факторами, и примерно 30% коек в детских стационарах заняты детьми с наследственной патологией [4]. Медицинские и социальные последствия этого заключаются в повышении потребности в медицинской помощи, сниженная продолжительность жизни больных, инвалидизация и большие экономические затраты на их содержание. С этих позиций необходимость профилактики ВПР и НЗ становится первостепенной задачей современного здравоохранения, т.к. подобная «генетическая отягощенность» популяции отражается на генофонде нации в целом. Регистрация частоты распространения таких аномалий, изучение молекулярных механизмов их патогенеза, использование медико-генетического консультирования, применение современных молекулярных методов диагностики и другие пути профилактики позволяют снизить частоту рождения детей с ВПР и НЗ на 60-70% [1]. Подобные работы необходимы для разработки мероприятий по снижению риска и уровня врожденных аномалий среди общей структуры населения.
Нами, в соответствии с правилами Европейского международного регистра (EUROCAT), проведены медико-генетические исследования среди населения трех крупных районов Западной зоны Азербайджана с целью дальнейшего составления регистра фенотипически наиболее легко диагностируемых ВПР и НЗ, подлежащих обязательной регистрации [5].
Материалы и методы исследований
Материал собран в экспедиционных условиях в селах и в районных центрах Таузского, Казахского и Акстафинского районов Западной зоны республики с 2005 по 2007 гг. Использованы списки ВТЭК ЦРБ для выявления больных с врожденной и наследственной патологией. В селах при подворовом обходе семей пробандов составлены родословные, и путем генеалогического анализа дифференцированы случаи ВПР и НЗ. Фенотипические частоты выявленных врожденных и наследственных патологий определяли по методике Ли, 1978 [3]. В качестве материала для анализов использовали образцы крови, забор которой производили из пальца в микропробирки с антикоагулянтом (гепарин или натриевая соль этилендиаминтетраацетата). С целью выявления β-талассемии и недостаточности фермента глюкозо-6-фосфатдегидрогеназы среди школьников использованы скрининг-программы [7]. Для идентификации типа мутации β-талассемии использован молекулярный метод высокотемпературной аллель-специфической амплификации, основанный по принципу метода полимеразно-цепной реакции [6,10]. Для выявления наследственных гемоглобинопатий использован метод электрофореза гемоглобина на ацетат-целлюлозных пленках и аналитический метод изоэлектрофокусирования гемоглобинов в полиакриламидно-амфолиновых пластинках c рН 3,5-9,5 [8,9].
Результаты исследований
Анализ полученных данных по распространению ВПР и НЗ в трех районах Западной зоны с населением 345980 человек показал высокую частоту встречаемости изученных патологий (рис.1). Так, во всех трех районах установлено по 9 патологий, представленные в Таузе (I) 20-ю формами с фенотипической частотой 0,0014 - 0,0650%, в Казахе (II) - 22-мя с частотой 0,0008 - 0,0301%, в Акстафе (III) - 19-ю нозологическими формами с частотой в пределах 0,0013 - 0,0172%. Наиболее распространенными заболеваниями здесь являются нарушения ЦНС (26,34%, 36,79%, 33,56%), врожденные аномалии слуха (42,47%, 26,42%, 27,52%) и аномалии зрения (8,87%, 12,26%, 11,41%, соответственно). Среди патологий ЦНС выявлено до 9 клинических форм, в том числе олигофрения, энцефалопатия, ДЦП и Spina bifida. В I и II районах среди мальчиков до 14 лет зафиксировано несколько случаев заболеваемости синдромом Клайнфелтера. Во всех трех районах частота глухоты и тугоухости превалирует у детей. Частота остальных патологий распределена следующим образом: врожденные пороки сердца - 4,84, 6,92 и 4,70%; аномалии скелета - 5,65, 5,66 и 5,37%; расщелина губы/неба - 5,11, 5,35 и 6,04%, соответственно.
Установлено, что среднее значение фенотипических частот пороков по трем районам следующее: в случае врожденных аномалий слуха - 0,0192%, гемолитической болезни - 0,0141%, аномалий скелета - 0,0132%, врожденных пороков сердца - 0,0131%, расщелины губы/неба - 0,0129%, нарушений ЦНС - 0,0099%, врожденных патологий зрения - 0,0098%, гемофилии - 0,0036% и большой талассемии - 0,0012% (табл.1).
Моногенные заболевания, выявляемые биохимическими методами, представлены гемолитической болезнью - 4,84, 5,03 и 8,72%; гемофилией - 1,34, 1,26 и 2,01%; большой талассемией - 0,54, 0,31 и 0,67%, соответственно. В I районе из 18-ти детей родившихся с диагнозом гемолитическая болезнь нами у 9-ти мальчиков и двух девочек определена активность фермента глюкозо-6-фосфатдегидрогеназы (Г6ФД). У восьми мальчиков, являющихся гемизиготами по дефициту фермента, удалось обнаружить полный и частичный дефицит Г6ФД. Трое имели нулевую активность Г6ФД «0». Пятеро мальчиков имели частичный дефицит фермента с фенотипом - Г6ФД «+». У разнополых больных детей с клиническим диагнозом большая талассемия идентифицирован тип мутации β-глобинового гена. Больной шести лет с клиническим диагнозом большая талассемия являлся гомозиготным по одной мутации - замена нуклеотида гуанин на нуклеотид аденин в 110-й позиции малого - первого интрона β-глобинового гена с генотипом - β+-IVS-1-110 (G-A)/ β+-IVS-1-110 (G-A).
Таблица 1. Фенотипические частоты патологий, установленных в Западной зоне республики
|
Патологии |
Фенотипические частоты (среднее значение), % |
||
|
Тауз |
Казах |
Акстафа |
|
|
Нарушение ЦНС |
0,0095 |
0,0106 |
0,0095 |
|
Врожденные пороки сердца |
0,0122 |
0,0179 |
0,0093 |
|
Аномалии скелета |
0,0142 |
0,0147 |
0,0106 |
|
Врожденные аномалии слуха |
0,0268 |
0,0171 |
0,0136 |
|
Гемолитическая болезнь |
0,0122 |
0,0130 |
0,0172 |
|
Гемофилия |
0,0034 |
0,0033 |
0,0040 |
|
Большая талассемия |
0,0014 |
0,0008 |
0,0013 |
|
Врожденные пороки зрения |
0,0075 |
0,0106 |
0,0113 |
|
Расщелина губы/нёба |
0,0129 |
0,0138 |
0,0119 |
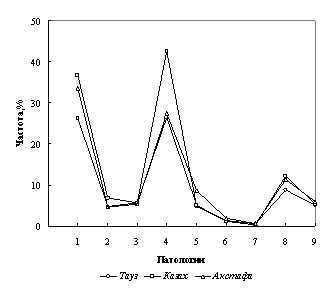
Рис. 1. Частота ВПР и НЗ в Западной зоне Азербайджана (1. Нарушение ЦНС; 2. Врожденные пороки сердца; 3. Аномалии скелета; 4. Врожденные аномалии слуха; 5. Гемолитическая болезнь; 6. Гемофилия; 7. Большая талассемия; 8.Врожденные пороки зрения; 9. Расщелина губы/нёба)
У девочки трех лет диагностированы две мутации: замена нуклеотида гуанин на нуклеотид аденин в 110-й позиции малого - первого интрона β-глобинового гена с генотипом - β+-IVS-1-110 (G-A) и замена нуклеотида гуанин на нуклеотид аденин в первой позиции большого второго интрона β-глобинового гена с генотипом - β0-IVS-2-1 (G-A). Т.о. компаундный генотип пробанда следующий: β+-IVS-1-110 (G-A)/ β0-IVS-2-1 (G-A). Во II районе среди 16-ти детей с диагнозом гемолитическая болезнь, у 9-ти (мальчиков - 6, девочек - 3) проведен анализ определения активности фермента Г6ФД. У пяти из шести мальчиков установлен дефицит фермента Г6ФД с нулевой активностью. Девочки имели дефицит фермента с гетерозиготным генотипом. У одного больного с диагнозом большая талассемия удалось установить тип мутации в бета-глобиновом гене. Клинически соответствующий большой талассемии больной имел компаундный генотип с двумя различными мутациями: замена нуклеотида гуанин на нуклеотид аденин в 110 позиции первого интрона (IVS-1-110, G-A) и замена нуклеотида гуанин на аденин в первой позиции второго интрона бета-глобинового гена (IVS-II-1,G-A). Т.о. генотип больного с диагнозом большая талассемия - IVS-1-110, G-A / IVS-II-1,G-A. В III районе из 13-ти детей с диагнозом гемолитическая болезнь у 5-ти мальчиков удалось обнаружить полный и частичный дефицит фермента глюкозо-6-фосфатдегидроге-назы. Все больные являлись гемизиготами по дефициту фермента. Трое мальчиков имели частичный дефицит фермента с фенотипом - Г6ФД «+». У двух мальчиков наблюдали нулевую активность фермента - Г6ФД «0». У одного мальчика в возрасте 2,5 лет выявлена большая талассемия. Удалось идентифицировать тип мутации β-глобинового гена. Больной являлся гомозиготой по одной мутации - замена нуклеотида гуанин на нуклеотид аденин в 110-й позиции малого - первого интрона β-глобинового гена с генотипом - β+-IVS-1-110 (G-A)/ β+-IVS-1-110 (G-A).
Таким образом, установление регионов с повышенным уровнем врожденных заболеваний, выяснение природы их наследования, точное определение нозологического диагноза, идентификация типов мутаций наиболее характерных для популяций патологий необходима для составления государственного регистра ВПР и НЗ, проведения квалифицированного медико-генетического консультирования. Подобные мероприятия, в конечном итоге, в будущем позволят снизить груз патологической отягощенности популяций и предотвратить рождение детей с врожденными и наследственными патологиями.
СПИСОК ЛИТЕРАТУРЫ:
- Бочков Н.П. Клиническая генетика: Учебник. - 2002. - М.: ГЭОТАР-МЕД. 448 с.
- Вахитов В., Викторова Т. Воздействие внешней среды на генетику человека. http://vatandash.ufanet.ru/07_04/72.htm
- Ли Ч. Введение в популяционную генетику. - 1978. - М.: Мир. 546 с.
- Медицинская Компания ИДК. Скрининг врожденных пороков развития. http://hello.mc-idk.ru/spec-articles.php
- Пузырев В.П., Эрдыниева Л.С., Кучер А.Н., Назаренко Л.П. Генетико-эпидемиологическое исследование населения Тувы. - 1999. - Томск: STT. 256 с.
- Шостакович-Корецкая Л.Р., Маврутенков В.В., Братусь Е.В., Маврутенкова Т.В. Полимеразно-цепная реакция: принципы и практические рекомендации по использованию в клинической практике врача (руководство для врачей). - 2002. Днепропетровск: МЗ Украины. http://savon.com.ua/diagnostic3.html
- Modell B. The ethics of prenatal diagnosis and genetic counseling // World Health Forum. - 1990. - 11. P. 179.
- Morengo-Rowe A.J. Rapid electrophoresis on cellulose acetate // J. Clin. Pathology. - 1965. - 18. P. 790.
- Rasulov E. Express-methods of hemoglobinopathy diagnosis in newborns // Journal of molecular genetics, microbiology, virusology. - 1990. - 1. P.27.
- Saiki R. K., Scharf S., Faloona F., Mullis K. B., Horn G. T., Erlich H. A., Arnheim N. Enzymatic amplification of beta-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia // Science. - 1985. - 230. P. 1350.
Библиографическая ссылка
Акперова Г.А. ИССЛЕДОВАНИЕ ВРОЖДЕННЫХ ПОРОКОВ РАЗВИТИЯ И НАСЛЕДСТВЕННЫХ ЗАБОЛЕВАНИЙ В ЗАПАДНОЙ ЗОНЕ АЗЕРБАЙДЖАНА // Успехи современного естествознания. 2007. № 10. С. 22-25;URL: https://natural-sciences.ru/en/article/view?id=11556 (дата обращения: 02.07.2026).