


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
USAGE OF MEK INHIBITORS IN ONCOLOGY: RESULTS AND PERSPECTIVES

Сокращение: НМРК – немелкоклеточный рак легкого, МАРК – митогенактивированные протеинкиназы, FDA(Food and Drug Administration) – Управление по санитарному надзору за качеством пищевых продуктов и медикаментов, ЦНС – центральная нервная система.
Митогенактивированные протеинкиназы (МАРК) – это семейство энзимов, которые отвечают за трансдукцию сигнала у всех эукариотов и осуществляют связь между внеклеточными сигналами и внутриклеточными путями экспрессии генов. Эти протеинкиназы регулируют и активируют одна другую с помощью системы фосфорилирования. МАРК контролируют множество разнообразных физиологических процессов макроорганизмов. Известны 3 подсемейства МАРК. Киназы, регулируемые внешними сигналами (ERK), отвечают за клеточное деление. Аминоконцевые киназы С-Jun (JNK) являются очень важными регуляторами процесса транскрипции. МАРКиназы р38 активируются с помощью воспалительных цитокинов и сигналов из окружающей среды при стрессе. Киназы JNK и р38 запускают апоптоз [89]. Особую роль в пролиферации клетки и карциногенезе играет путь МАРК RAS/RAF/MEK/ERK. Он представляет собой типичный каскад и включает цепь протеинов и киназ, включая белок саркомы крыс (RAS), митоген активированную протеинкиназу киназы киназы (RAF или МАР3К), митоген активированную протеинкиназу киназы (МЕК или МАР2К) и протеинкиназу, регулируемую внешними сигналами (ERK или МАРК). Этот МАРК каскад (RAS/RAF/MEK/ERK путь) является путем, который регулирует клеточную пролиферацию, клеточный цикл и миграцию клетки (рис. 1). При развитии рака у человека мутации семейства RAS/RAF наиболее часто являются причиной нарушения регуляции трансдукции сигнала через этот путь. Онкогенная активация ras встречается при 30 % опухолей человека [89]. Мутации КRAS обнаружены в 90 % случаев рака поджелудочной железы, 40–50 % рака толстой кишки и щитовидной железы и в 20 % – немелкоклеточного рака легкого [55, 75, 44]. Мутации в BRAF были обнаружены у более чем 60 % больных меланомой [90, 21] и от 40 до 60 % больных папилярным раком щитовидной железы [20, 24, 113, 63], 20–33 % низкодифференцированным раком яичников [98, 112], 5–8 % колоректальным раком [25], у 2–5 % – немелкоклеточным раком легкого (88, 16). Хотя мутации МЕК 1/2 обнаруживают редко, постоянно активный МЕК был обнаружен в исследованиях более чем в 30 % клеток первичной опухоли [46].
Передача сигнала МЕК. К настоящему времени определены 4 отличных друг от друга пути МАР киназ, в которых представлено 7 МЕК энзимов (рис. 1).
МЕК 1 и 2 являются типичными протеинами подсемейства МЕК [3]. Путь RAS/RAF/MEK/ERK активируется широким спектром ростовых факторов и цитокинов, действующих через рецепторы тирозин-киназ. Ростовые факторы ЕGF, IGF и TGF сначала связываются, а затем активируют трансмембранные рецепторы, расположенные на поверхности клетки. Активированные рецепторы далее связывают множество белков-передатчиков, которые в свою очередь вовлекают белки обмена нуклиотидов. Белки обмена активируют RAS путем превращения связанных форм из неактивной GDP в активную GTР форму. Активированный RAS запускает RAF киназу в мембране, где ряд фосфорилирующих реакций также приводит ее в активное состояние. Активированный RAF форсфорилирует и активирует МЕК-киназу. МЕК-киназа в свою очередь фосфорилирует и активирует ERK-киназу. Фосфорилированная ERK может перемещаться в ядро, где она фосфорилирует и активирует ряд факторов транскрипции [22, 110, 103]. Это приводит к изменению транскрипции гена и пролиферации и дифференцировке клетки [61, 82, 76, 108]. Семейство протеинкиназ RAF состоит из А-RAF, В-RAF и С-RAF (RAF-1). Для всех этих киназ RAS является общим активатором, МЕК 1/2 – основным эффектором [39]. МЕК 1/2 являются киназами с двойной специфичностью (обладают двойным специфическим действием) и катализируют фосфорилирование тирозина и треонина на ERK1 и ERK2, которые известны как их единственные физиологические субстраты [95]. В противоположность RAF и МЕК 1/2, у которых субстрат узко специфичен, активированные ERK 1/2 катализируют фосфорилирование множества субстратов цитоплазмы и ядра, регулирующих различные функции клеток: митоз, эмбриогенез, клеточную дифференцировку, подвижность, метаболизм и программированную гибель, а также ангиогенез (рис. 2) [54, 4, 23, 66, 80, 91, 97, 25, 83].
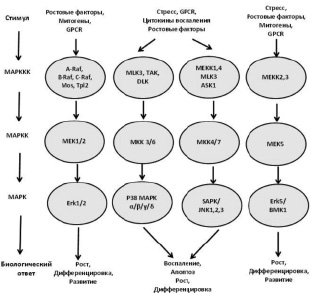
Рис. 1. MAPK каскады у млекопитающих (иллюстрация воспроизведена с сайта Cell Signaling Technology, Inc. (www.cellsignal.com). Примечание. GPCR – рецептор, соединенный с G – протеином
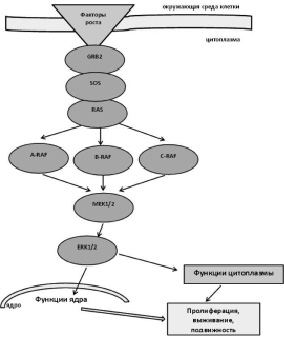
Рис. 2. Сигнальный путь RAS-RAF-MEK-ERK. Примечание. SOS (son of sevenless) – активированный гуанин нуклеотидный фактор обмена, приводящий к активированию RAS; Grb2 – связывающий протеин
МЕК ингибиторы и их механизм действия
Было показано, что аберрантная передача сигнала через RАS/RAF/МЕК-путь приводит к трансформации клетки [61]. Например, активация МАРК важна для генной регуляции в фазе G1 клеточного цикла перед репликацией ДНК, а также при сборе веретена во время клеточного деления путем мейоза и митоза. Характерным свойством многих опухолей является нарушенная активация пути МАРК вследствие мутаций белков, вызванных онкогенами EGFR, PI3K, RAS, RAF. Поэтому, ингибирование МЕК1/2 является перспективным подходом к блокированию каскада сигналов RAF/RAS. Характеристики структуры МЕК1/2 также имеют уникальное преимущество для использования этой молекулы в качестве таргетной. Она имеет карман, смежный с сайтом для связывания Мg АТФ, который сохранился только в МЕК-протеинах. При связывании ингибитором происходит последовательное изменение конформации, приводящее к закрытию нефосфорилированного МЕК1/2 и каталитически неактивному состоянию. Механизм неконкурентного связывания АТФ не вызывает ингибирования благодаря уникальному карману АТФ. Это позволяет избежать нежелательных побочных эффектов, связанных с необратимым ингибированием других протеинкиназ, и не вызывает конкурирование с внутриклеточными концентрациями АТФ, что могло бы создавать ряд проблем. Иными словами, гидрофобный аллостерический карман, смежный с АТФ связывающим сайтом, – уникальное отличие МЕК от других киназ, позволяет селективно ингибировать другой сайт, а не высокоспецифичную АТФ этой зоны [87, 104, 81]. Были разработаны несколько соединений с высокой ингибиторной активностью, исключительно действующих на МЕК1/2, которые были исследованы в клинических испытаниях.
Все семь идентифицированных энзимов МЕК-семейства селективно фосфорилируют серин/треонин- и тирозин-окончания их таргетов, вниз по ходу трансдукции сигнала по каскаду МАРК. В общем, белки МЕК имеют схожую структуру, которая включает домен с амино-концом, каталитический домен (домен киназы) и домен с карбокси-концом. Отличительной особенностью каждого из МЕК-белков являются их индивидуальные концевые последовательности. Вместе с тем МЕК1 и МЕК2 крайне важные медиаторы RAS/RAF/МЕК/ЕRК-пути, имеют относительно близкую структуру и функцию. Последовательность N-конца имеет док-сайт для субстрата ЕRК, последовательность для экспорта в ядро, уникальную для МЕК1/2, и ингибиторный/аллостеричный сегмент. Домен протеинкиназ высокоспецифичен и содержит главный каталитический сайт и сайт связывания АТФ, который находится возле ингибиторного/аллостерического сегмента в пределах N-конца. С-конец включает в себя домен для универсального дока, связывающий сайт для ближайших компонентов вверх по каскаду – Rаf-киназы.
МЕК-ингибиторы представляют собой малые молекулы, которые ингибируют МЕК-форсфорилирование. Для этого МЕК-ингибиторы связываются с ингибиторным/аллостерическим сегментом смежным с АТФ-связывающим сайтом, вмешиваясь неконкурентно в работу с помощью протеинкиназ. Уникальный связывающий сайт МЕК-ингибиторов позволяет благодаря высокой специфичности подойти только к МЕК-протеинам и препятствует перекрестному ингибированию других серин/треониновых протеинкиназ. Все это приводит как к снижению активности МЕК, так и количественному уменьшению активированной ЕRК в клетке. Используемые в настоящее время в клинических испытаниях ингибиторы МЕК являются препаратами для приема внутрь один или два раза в день. Их метаболизм осуществляется системой цитохромов р450 в печени [81].
Эффективность ингибиторов МЕК
В настоящее время в клинических исследованиях изучают несколько ингибиторов МЕК. Однако эти препараты существенно отличаются по их фармакокинетике и способности устойчиво ингибировать МЕК-зависимую активацию ЕRK.
Первый ингибитор МЕК СI-1040 был включен в I фазу клинических испытаний в 2000 г., но к настоящему времени только один ингибитор МЕК был одобрен FDA. Это связано с тем, что хотя и имелись некоторые данные об активности препаратов, при большинстве видов опухолей она была незначительной.
Использование МЕК-ингибиторов в качестве монотерапии продемонстрировали свою эффективность при наличии BRAF- и NRAS-мутаций, а при опухолях с КRAS мутациями их эффективность была непостоянной. В настоящее время имеются исследования по синергическому действию комбинаций МЕК-ингибиторов с RAF, РI3К и АКТ-ингибиторами, а также их сочетанию с различными цитостатиками (гемцитабином, таксанами и др.). Доклинические исследования показали противоопухолевую эффективность ингибиторов МЕК при меланоме, раке толстой кишки, раке молочной железы, немелкоклеточном раке легкого, поджелудочной и щитовидной желез.
Токсичность ингибиторов МЕК
После исследования большого количества МЕК-ингибиторов у больных раком появились данные о токсичности, связанные с механизмом их действия. Большинство из них является общими для всех малых молекул ингибиторов киназ – сыпь, усталость и диарея. Сыпь является одним из наиболее частых и дозолимитирующих побочных эффектов этой группы препаратов. В некоторых исследованиях ее частота достигает 80 %. Было установлено, что в основе этой кожной токсичности лежит ингибирование RAF/MEK/ERK в МАРК пути в кератоцитах на уровне EGFR или ниже, на уровне МЕК [49]. Однако нет данных о том, что она коррелирует с эффективностью, как это отмечено с таргетной терапией EGFR блокаторами [68]. Специфичная токсичность для МЕК ингибиторов включает нарушения зрения в виде помутнения и потерю остроты зрения. Есть описание окклюзии вен сетчатки [72], но наиболее часто встречается тяжелая центральная ретинопатия [29]. Эта токсичность носит обратимый характер после снижения дозы или отмены препаратов. Кроме того, отмечались периферический отек, частичный периорбитальный отек, а также высокий уровень креатинфосфокиназы, не связанный с выходом за пределы нормы тропонина при отсутствии рабдомиолиза или за счет другой патологии, а также редкие случаи дисфункции левого желудочка или влияние на ЦНС, включая галлюцинации и расстройства (предположительно связанные с хорошим проникновением в ЦНС).
Далее в статье представлены сведения, полученные в ходе клинического применения отдельных препаратов-ингибиторов МЕК.
CI 1040 (РD184352) был первым аллостерическим ингибитором МЕК1/2, который использовали в клинических испытаниях. В I фазе клинических исследований наиболее частой токсичностью была сыпь, астения, диарея, тошнота и рвота. Использование пищи с высоким содержанием жира усиливало действие препарата. Биопсии 10 больных, взятые до и после лечения, в дозе, рекомендованой для II фазы, показали ингибирование фосфорилирования ЕRК в среднем до 75 % (46–100 %) [71]. Этот препарат был рекомендован для исследований II фазы для лечения больных немелкоклеточным раком легкого, рака молочной железы, толстого кишечника, поджелудочной железы, однако из 67 больных, включенных в исследование, объективного эффекта не отмечено ни у одного [94]. Данные фармакокинетики очень разнились между больными. Исследование этого препарата послужило основой для разработки соединения с улучшенными фармакологическими свойствами (РD-0325901).
РD-0325901(AS703026, MSC1936369B) является неконкурентным АТФ ингибитором МЕК 1/2. Этот препарат был разработан как аналог CI 1040. Он имеет улучшенную биодоступность при приеме внутрь и значительно более высокий потенциал против МЕК1/2. В I фазе клинических исследований была доказана его противоопухолевая активность у больных с меланомой, однако, включение больных в исследование было прекращено в связи с наличием побочного действия в виде окклюзии вен сетчатки [95]. II фаза исследования предварительно леченных больных НМРЛ не достигла своей конечной цели по эффективности [42]. Недавнее исследование РD-0325901 в сочетании с двойным ингибитором PI3К/mТОR РF04691502 выявило токсичность, которая препятствовала назначению препарата в эффективных дозах [15]. В настоящее время продолжают проводить ряд исследований по его применению совместно с ингибиторами PI3К/mТОR [81].
Selumetinib (АZD6244). Селуметиниб – потенциальный, неконкурентный АТФ ингибитор, имеющий высокую специфичность к МЕК 1/2 по сравнению с другими протеинкиназами. Селуметиниб является, пожалуй, самым широко изученным ингибитором МЕК в клинике. В испытаниях I фазы эритематозная макулопапиллярная сыпь была наиболее частой и дозолиметирующей токсичностью и отмечалась в 74 % случаев, 3–4 степень при этом была отмечена в 20 % случаев. Кроме того, у больных встречались диарея (56 %) и инверсия Т-зубца при гипоксии (1 больной). После 2-х циклов лечения у 19 из 57 больных (33 %) достигнута стабилизация. Девять больных (16 %) имели стабилизацию длительностью 5 мес., а двое до 19 и 22 мес. [1]. В 19 парных пробах биопсии опухолей было отмечено ингибирование форсфорилирования ЕRК на 79 % в среднем. В одном случае с медулярным раком щитовидной железы, а также у одного больного с меланомой радужки и раком почки была достигнута длительная стабилизация заболевания [1]. Соединение было оформлено в капсулу для приема внутрь с улучшенными фармакологическими свойствами. В I фазе испытания отмечался длительный положительный ответ на лечение у больного с меланомой с BRAFV600Е-мутацией [6].
Монотерапия селуметинибом оценивалась в нескольких исследованиях II фазы при различных солидных опухолях и гематологических заболеваниях [86, 9, 43, 85, 17]. Лечение селуметинибом 28 больных метастатическим раком желчевыводящих путей сопровождалось объективным ответом у 3-х больных (12 %) и стабилизацией у 14 больных, вместе с тем при папиллярном раке щитовидной железы, как и при гепатоцеллюлярной карциноме никакой клинически значимой противоопухолевой активности не было отмечено. В исследовании меланомы с мутацией BRAF V600К у 10 больных не отмечалось никакого противоопухолевого эффекта при наличии высокого уровня фосфорилирования АКТ (фАКТ), наряду с этим у 3-х из 5 больных с низким уровнем фАКТ в меланоме была достигнута регрессия опухоли, что заставляет предположить потенциальную роль активации РI3/АКТ в резистентности ингибиторов МЕК. Незначительный противоопухолевый эффект был отмечен при рецидивах или рефракторном остром миелолейкозе.
Селуметиниб далее также оценивали в комбинации с другими противоопухолевыми препаратами. В исследовании I фазы комбинации селуметиниба с ингибитором АКТ МК-2206 дозолиметирующей токсичностью являлись сыпь, стоматит, отслойка пигментного эпителия сетчатки 2 ст., диарея, повышение липазы 4 степени, двусторонняя катаракта 1 степени, слабость. В этом исследовании с участием 51 больного длительный подтверждаемый частичный ответ был отмечен у 1-го больного НМРЛ с КRAS-мутацией и у 1-й больной раком яичников с КRAS-мутацией, а неподтвержденный длительный частичный эффект наблюдался у 1 больного раком поджелудочной железы [57]. В рандомизированном исследовании II фазы, в котором оценивалась эффективность доцетаксела с солументинибом или с плацебо у предварительно леченных больных НМРЛ с KRAS-мутацией, в группе с селуметинибом достигнута статистически значимая разница в безрецидивной выживаемости – 5,3 мес. против 2,1 мес. при использовании плацебо (р ≤ 0,014). Общая выживаемость статистической разницы в этих группах не достигла, хотя имела тенденцию к улучшению [53]. В исследовании I фазы, оценивающем комбинацию селуметиниба и цетуксимаба при солидных опухолях и колоректальном раке с КRAS-мутациями, наиболее частой токсичностью являлись акнеподобная сыпь, слабость, тошнота/рвота, диарея. Дозолимитирующая токсичность была представлена гипомагнезиемией 4 степени [27]. Из 13 больных, которым проводилось лечение с эскалацией дозы, частичный эффект отмечен у 2-х больных колоректальным раком, у 1-го больного плоскоклеточным раком миндалин, у 1-го – НМРЛ и у 2-х – колоректальным раком. Рандомизированные исследования II фазы, сравнивающих селуметиниб с темозоламидом при лечении меланомы в I линии, с пеметрекседом при НМРЛ после I–II линии терапии, с капецитабином при раке поджелудочной железы после прогрессии на гемцитабине, с капецитабином при колоректальном раке после I–II линии терапии не продемонстрировало преимуществ, хотя в каждом из исследований отмечалась противоопухолевая активность препарата в монотерапии [40, 11, 12, 59]. В настоящее время проводятся исследования I–II фаз сочетания селуметиниба с различной таргетной терапией, например, препаратом саракатинибом (АZD0530) [30] или ХТ с использованием вандетаниба, доцетаксела, гемцитабина, иринотекана, циклоспорином (модулятором Wnt (кальциевого пути)) [102], фенформином [115].
Траметиниб (GSK 1120212, JTP-74057) – это малая молекула, аллостерический неконкурентный АТФ ингибитор МЕК. В доклинических испытаниях, проведенных на линии колоректального рака и на моделях ксенографта, он показал потенциальную активность ингибирования ЕRК в клетках с мутациями BRAF или KRAS [114].
В I фазе исследования траметиниба участвовало 206 больных [29]. Наиболее часто встречалась сыпь и диарея, а дозолимитирующей токсичностью, кроме последних, была центральная серозная ретинопатия. Эффективный период полураспада траметиниба составляет около 4 дней. Хотя максимально переносимой дозой была доза 63 мг, во II фазе была рекомендована ежедневная доза 2 мг в связи с плохой переносимостью после первого курса лечения. При всех уровнях дозы объективные эффекты отмечались в 10 %, причем наибольшая чувствительность к лечению отмечена при меланоме с BRAF-мутацией [49]. Falchook с соавт. провели анализ подгрупп больных с меланомой с BRAF-мутацией V600E или V600K, в котором выявили ответ у 33 % больных, не получавших ингибиторы BRAF. Далее этот результат был подтвержден во II фазе, в которой эффект был получен у 25 % больных, не получавших ингибитор BRAF, тогда как при резистентном к ингибиторам BRAF варианте заболевания отмечена минимальная клиническая активность [58, 36]. В III фазе больных с меланомой с BRAFV600Е/К, которых ранее не лечили ингибиторами BRAF или МЕК или ипилимумабом, рандомизировали в группы – для лечения траметинибом или химиотерапией дакарбазином или паклитакселом. В этом исследовании, пациенты в рукаве с траметинибом имели медиану выживаемости без прогрессирования – 4,8 мес., что на 1,5 мес. статистически достоверно было больше, чем в группе с ХТ, а общая выживаемость после 6 мес. лечения была 81 % и 67 % соответственно (НR = 0,54, р ≤ 0,01) [32]. В мае 2013 г. FDA одобрило MekinistTM (траметиниб диметил сульфоксид, GlаxoSmithKline, LLC) для лечения больных с нерезектабельной или метастатической меланомой при наличии BRAF-мутаций.
Как показывают данные доклинических исследований, эффективность МЕК-ингибиторов может быть повышена ингибиторами RAF [35, 48]. Кроме того, активация пути МАРК была установлена как потенциальный механизм резистентности к ингибиторам RAF при меланоме.
Таким образом, одновременное ингибирование RAF и МЕК становится перспективным в качестве подхода, преодолевающего резистентность к ингибиторам BRAF. Была проведена I/II фаза исследования для определения безопасности и с целью сравнить дабрафениб (ингибитор BRAF), как монотерапию и в комбинации с траметинибом. Как и предполагалось, в рукаве с комбинацией было значительно меньше случаев плоскоклеточного рака кожи – побочного эффекта, связанного с BRAF-ингибированием, в основе которого лежит парадоксальная активация МАРК пути. Отмечено значительное повышение выживаемости без прогрессирования в рукаве с комбинированной терапией (р<0,001), что показывает потенциал применения ингибиторов МАРК в замедлении процесса развития резистентности к BRAF ингибированию [69].
Следует отметить, что количество ответов на траметиниб у больных меланомой с ВRАF-мутациями меньше, чем при использовании ингибиторов ВRАF (вемурафениб, дабрафениб). Этот факт требует объяснения, так как основным субстратом по пути вниз от ВRАF является МЕК.
Изучались комбинации с траметинибом и при других видах рака. В рандомизированном плацебо контролируемом исследовании, включавшем 160 больных метастатическим раком поджелудочной железы независимо от статуса мутаций КRAS, сочетание траметиниба и гемцитабина не привело к повышению эффективности лечения [51]. Продолжаются другие исследования I/Ib фазы комбинаций траметиниба с различными таргетами и химиопрепаратами (эверолимус, пазопаниб, дабрафениб, эрлотиниб, фторурацил), а также с лучевой терапией, ингибитором АКТ (GSK2141795) и ингибитором РI3К (ВКМ120) [50, 62, 2, 8].
Кобиметиниб (GDC-0973, XL-518, RG-7421) – это неконкурентный ингибитор МЕК 1/2, показавший свою активность в клеточных линиях с мутациями KRAS и BRAF [111].
Предварительные исследования I фазы показали, что больные хорошо переносили этот препарат. Была получена длительная стабилизация заболевания у больного НМРЛ [34]. Далее это соединение изучалось в исследовании Ib фазы в комбинации с препаратом, направленным на РI3К, для лечения генерализованных солидных опухолей. При этой комбинации наиболее частыми побочными эффектами являлись диарея, усталость, рвота и кожная сыпь. Также были отмечены повышение уровня липазы 3 степени и креатинфосфокиназы, что являлось дозолимитирующей токсичностью. Среди 46 больных частичная регрессия была отмечена у 1-го больного меланомой с BRAF-мутацией, 1 больного раком поджелудочной железы с BRAF-мутацией, и у 1 больного раком эндометрия с КRAS-мутацией [96, 70]. В другом исследовании Ib фазы GDC-0973 было сочетание с вемурафенибом при меланоме с BRAFV600 мутацией. Согласно предварительным исследованиям уменьшение размеров опухоли было достигнуто у всех 8 больных, не получавших ранее вемурафиниб. Интересно то, что только у одного из 44 больных был выявлен плоскоклеточный рак кожи [37]. На основании этих результатов была начата III фаза исследований, в которой сравнивают эту комбинацию с вемурафенибом.
Продолжаются исследования сочетания GDC-0973 с другими таргетными препаратами (ингибитором РI3К (GDC-0941) и ингибитором АКТ (GDC-0068).
Пимазертиб (АS703026, МSС 1936369В) – это неконкурентный АТФ ингибитор МЕК1/2. Пимазертиб оценивали с I фазе исследования [26, 47]. Среди наиболее часто встречаемых побочных эффектов отмечена кожная сыпь, диарея, астения, анорексия, тошнота, рвота, периферические отеки, нарушение зрения и анемия. Дозолимитирующей токсичностью была окклюзия вен сетчатки 2 степени тяжести, повышение функциональных проб печени 3 степени тяжести, кожная сыпь, фарингит, акнеподобная сыпь, отслойка сетчатки и отек макулы. У 75 больных отмечено сокращение размеров опухоли, у всех из них были выявлены BRAF- или NRAS-мутации. В исследовании I–II фаз пимазертиб был комбинирован с 5-фторурацилом, лейковарином и иринотеканом (FОLFIRI) в качестве II линии лечения метастатического колоректального рака с КRAS-мутациями. Однако исследование не перешло во II фазу в связи с тем, что эффективные дозы пимазертиба были не достигнуты по причине токсичности [73]. Среди продолжающихся исследований: Ib фаза исследования сочетания пимазертиба и ингибитора РI3К/mTOR (SAR 245409) при солидных опухолях [7], при меланоме с мутациями NRAS, при аденокарциноме поджелудочной железы и у онкогематологических больных.
Рефаметиниб ВАY 86-9766 (RDEA 119) представляет собой дериват циклопропан-1-сулфонамида и является неконкурентным АТФ, высокоселективным аллостерическим ингибитором МЕК1/2 [52]. В доклинических исследованиях, рефаметиниб был активен по отношению к меланоме, раку толстой кишки, кожи, поджелудочной железы [18]. В I–II фазах наиболее частой токсичностью была сыпь, а клинический эффект был ограничен [38]
Как выявила I фаза испытаний ВАY 86-9766 хорошо переносится, наиболее часто отмечалась акнеподобная сыпь и гастроинтенстинальная токсичность. Из 53 больных, у 1 больного раком толстой кишки отмечена частичная регрессия и у 11 больных отмечена длительная стабилизация [109]. Во II фазе ВАY 86-9766 сочетали с сорафенибом при лечении больных гепатоцеллюлярным раком в качестве I линии терапии. Из 70 больных у 3-х отмечена частичная регрессия и у 25 длительная стабилизация. Однако у 25 больных отмечена токсичность 4 степени, связанная с приемом препарата, что потребовало коррекции дозы [67]. Продолжается исследование I/II фазы, оценивающее ВАY 86-9766 в комбинации с гемцитабином у больных метастатическим раком поджелудочной железы. По предварительным данным, отмечен приемлемый профиль токсичности [105, 93]. Продолжена I фаза исследования, в котором изучается комбинация ВАY 86-9766 и ингибитора РI3К ВАY 80-6946 [92].
Биниметиниб МЕК 162, ARRV-438162) – неконкурентный АТФ ингибитор МЕК ½. Препарат МЕК 162 был оценен в I фазе исследований на большом количестве больных метастатическим колоректальным раком и генерализованным раком желчного пузыря [10, 31]. Отмечен частичный ответ в одном случае из 26 больных раком желчного пузыря. В группе с колоректальным раком исследование продолжается. II фаза исследований МЕК 162 при метастатической меланоме с NRAS или BRAF-мутациями объективные ответы наблюдались у 20 % больных с меланомой с NRAS- мутацией и у 20 % больных меланомой с BRAF-мутацией [5, 33]. Ответ на лечение в случае предшествующего лечения ингибораторами BRAF не получен. Основываясь на перспективных результатах лечения больных меланомой с этой мутацией, дополнительно 70 больных меланомой с NRAS мутацией были включены для изучения препарата МЕК 162 в этой популяции. Также запланировано рандомизированное исследование у больных с меланомой и NRAS-мутациями. Продолжаются исследования комбинаций МЕК 162 с различными препаратами, такими как ингибиторы РI3К BYL719 [56], ингибиторы RAF киназ LGX 8189062 и RAF 265 и паклитаксела при меланоме и других солидных опухолях [100].
RO 4987655 (CH 4987655). I фаза клинических исследований препарата RO 4987655 (СН 4987655) была проведена у 49 больных. Установлено, что, несмотря на то, что пациенты были сильно предлеченные, в 21,1 % отмечалась клиническая польза, включая 2 частичных регрессии (одну подтвержденную и одну неподтвержденную), при этом токсичность была управляемой (сыпь отмечена у 91,8 %, гастроинтестинальные нарушения – у 69,4 %). Дозолимитирующей токсичностью являлись нарушения зрения (у 1 больного) и повышение уровня креатинфосфокиназы (у 3-х больных). Исследователи предполагают, что препарат имеет перспективы при лечении больных с опухолями, несущими RAS- и/или RAF-мутации. Из 38 больных меланомой у одного ответ был подтвержден и у одного не был. Продолжаются исследования в расширенных кагортах больных меланомой с BRAFV600- мутациями, НМРЛ с КRAS-мутациями, колоректальным раком с мутациями KRAS и/или BRAFV600 [65].
RO 5126766 – это первый высокоселективный двойной ингибитор RAF/МЕК [77]. В исследовании I фазы среди 52 больных у 2-х больных меланомой с BRAF-мутацией и у 1-го меланомой с NRAS-мутацией отмечен частичный эффект. Побочные эффекты были управляемы. Наиболее часто отмечались сыпь, диарея, нарушение зрения. Дозолимитирующей токсичностью явились помутнение зрения и повышение уровня креатинфосфокиназы [77, 45].
Другие ингибиторы МЕК
Продолжены исследования I фазы АZD 8330 (ARRV-424704), WX-554, Е 6201 и ТАК-733 [31, 74, 101, 19]. В I фазе исследования АZD 8330 у одного больного меланомой отмечен частичный эффект. Наиболее частой токсичностью, связанной с препаратом, были акнеподобный дерматит, слабость, диарея и тошнота. Дозолимитирующей токсичностью были изменение ментального статуса и сыпь [19]. Доклинические испытания выявили эффективность препарата E6201, который позволяет преодолеть приобретенную резистентность к ингибитору BRAF вемурафенибу и аллостерическому МЕК-ингибитору селуметинибу за счет связывания с МЕК1 вдали от точечной мутации С121S. Это приводит к тому, что мутация не оказывает влияние на ингибиторную активность МАРК пути [84]. Новый аллостерический (несвязывающий селективный) ингибитор МЕК 1/2 ТАК 733 показал противоопухолевые свойства в различных клеточных линиях меланомы (кожной и увеальной) с различными онкогенными мутациями [106]. Полученные данные позволяют продолжить дальнейшие исследования в этом направлении.
Обсуждение и перспективы
Все вышеизложенное свидетельствует о том, что в онкологии МАРК сигнальный каскад является важным путем, а МЕК, как его центральная киназа, является предметом для изучения в различных клинических исследованиях. Однако, несмотря на то, что при раке аберрации в передаче сигнала в этом каскаде обнаруживают часто и МЕК1/2 в этом пути занимает ключевое положение, а также несмотря на то, что ингибиторы МЕК имеют высокий потенциал и специфичность, их применение не показало высокой терапевтической активности. Большинство исследований представляют данные, в которых эффективность лечения сводится преимущественно к стабилизации заболевания. Этот феномен можно объяснить, если воспользоваться результатами исследования, проведенного на клеточных линиях с получением небольшой активности МЕК-ингибиторов при солидных опухолях, в результате чего был сделан вывод о том, что МЕК ингибиторы имеют цитостатический эффект, а не цитотоксический [60]. Иными словами, хотя была обнаружена таргетная супрессия в ткани опухоли, она не достигала уровня, необходимого для цитотоксического действия на опухоль.
Однако нельзя не учитывать и тот факт, что дозолимитирующие побочные эффекты ингибиторов МЕК являются одной из причин их недостаточного клинического эффекта. Дозирование ингибиторов МЕК очень ограничено токсичностью (диарея, сыпь и др.), которая значительно влияет на качество жизни пациентов. На самом деле Infante с соавт. [49] не определили максимально переносимую дозу траметиниба, поскольку эскалация дозы после первого цикла лечения (28 дней) была лимитирована токсическими эффектами, возникающими при более высоких дозах. Получается, что возможность ингибировать МЕК ограничена. Эта мысль приводит к основному вопросу, общему для всей таргетной терапии в онкологии: насколько и как долго должна быть ингибирована таргетная точка для получения максимального клинического эффекта? Возможно ли получить эффект при коротком воздействии более высоких доз? Очевидно, что необходимо продолжать в клинике исследования по изучению фармакодинамики составляющих этого каскада [78].
Кроме ограничения в дозах, другой причиной низкого ответа на МЕК-таргетные препараты является работа противодействующих сигнальных каскадов в качестве прямого ответа на ингибиторы МЕК [30]. Следует иметь в виду, что к развитию рака приводят нарушения регуляции во множестве сигнальных путей, а ингибирование только одного из них недостаточно для апоптоза и остановки роста опухоли. Исходя из этого, исследования, появившиеся недавно, изучают возможность комбинации МЕК-ингибиторов с другими таргетными препаратами, которые ингибируют дополнительно другие пути, например РI3К/mТОR для усиления их цитотоксического эффекта. Кроме сказанного ранее, наиболее частые звенья МАРК пути с мутациями RAS и RAF, очевидно, имеют другие мишени помимо МЕК, и вероятно альтернативные пути компенсируют эффекты ингибиторов МЕК. И наконец, хотя МАРК путь активируется во многих клетках опухоли, при некоторых неоплазмах его функционирование может и не являться столь необходимым для их роста и выживания. Отсюда вытекает идея необходимости разработки клинических тестов на мутации в МАРК каскаде для изучения возможного эффекта от МЕК 1/2 ингибиторов при лечении больных раком с или без мутаций в МАРК каскаде. Иными словами, необходимы предсказательные биомаркеры для определения опухолей, которые можно ингибировать с помощью МЕК.
Возвращаясь к вопросу о невысокой эффективности при использовании ингибиторов МЕК в клинике, еще одно объяснение этому можно найти, если учесть что МЕК путь активирован исключительно при наличии RAS/RАF-мутаций и их активации. Доклинические исследования определили ауторегуляторную замкнутую цепь (петлю) с отрицательной обратной связью между ERK и RAF, которая передает чувствительность к МЕК-ингибиторам. Активированный ERK приводит к тоническому ингибированию RAF киназ, а далее к активированию RAF, что, в свою очередь, запускает антиапоптический каскад ниже RAF, тем самым нивелируя цитотоксическую активность МЕК-ингибиторов. Не менее интересно, почему ингибиторы МЕК эффективны при опухолях с ВRАF-мутациями? ВRАF является треонин/серин-содержащей киназой вниз по ходу от КRАS и вверх по ходу от МЕК. Хотя 3 RAF изоформы имеют аналогичную структуру, они имеют различную способность к фосфорилированию и активации МЕК, с преобладающей активностью киназ, относящейся к ВRАF. Это имеет клиническое значение, поскольку было установлено, что опухоли с мутациями ВRАF имеют отличительную способность к ответу на применение ингибиторов МЕК [99]. Опухоли с BRAF-V600E мутациями не имеют замкнутой цепи с отрицательной обратной связью, о которой говорилось выше, и являются чувствительными к ингибиторам МЕК [48]. Эти данные предполагают, что комбинация ингибиторов МЕК и RAF обладает синергизмом. Их сочетание с BRAF-ингибиторами может быть перспективным и привести к повышению частоты ответов и увеличению их длительности по сравнению с монотерапией BRAF-ингибиторами. Это предположение было подтверждено и в клинической практике, о чем говорилось ранее в статье.
Возможность получения ответа при совместном ингибировании ВRАF и МЕК может быть обусловлена тем, что ВRАF- ингибиторы путем промоции парадоксальной активации в нормальных тканях могут уменьшить токсические эффекты МЕК ингибиторов, что является основным моментом, ограничивающим дозы при использовании МЕК-ингибиторов и обеспечить введение эффективных доз для получения клинических ответов [78].
Также из исследований видно, что к ингибиторам МЕК имеется дифференцированная чувствительность. Хотя была показана активность в случае монотерапии меланомы с BRAF-мутацией, эта активность все-таки была несколько ниже таковой по сравнению с селективными BRAF-ингибиторами (вемурафенибом, добрафенибом). Кроме того, эти препараты не активны при опухолях с BRAF-мутациями в случае развития резистентности к ингибиторам BRAF. Но самое главное то, что ВRАF-мутированные клетки имеют большую чувствительность к трансдукции сигнала, чем RАS-мутированные клетки [99]. После открытия того, что гены, кодирующие RAS и изоформу RAF – ВRАF, являются онкогенными, было предпринято много усилий для доказательства гипотезы, которая предполагала, что раковым клеткам для роста и выживания необходимо наличие онкогена, или cостояние так называемой онкогенной зависимости. Данные доклинических исследований показывают, что активность протеинкиназ МЕК является необходимым условием для того, чтобы RAS и ВRАF запускали процессы клеточной пролиферации и способствовали выживанию, т.е. обусловливают состояние онкогенной зависимости [99, 28]. Следовательно, ингибирование МЕК может значительно повлиять на эти процессы у пациентов с RAS и ВRАF-мутациями через управление каскадом, так как КRАS и ВRАF активирующие мутации запускают канцерогенез через ключевую активацию МАРК пути [41].
Не исключено, что применение ингибиторов МЕК при опухолях с NRAS мутациями может также оказаться перспективным. Falchook с соавт. обратили внимание на некоторую активность трамитиниба при RАS-мутированных раках [49, 29]. В ситуациях с RAS-мутированными опухолями именно их комбинации с другими препаратами могли бы усиливать ингибирование каскада [79]. Здесь нельзя не упомянуть еще одну причину, которая делает привлекательным применение МЕК-ингибиторов в клинике. Поскольку RAS и RAF-мутации могут привести к постоянно активному состоянию ЕRK, ингибирование МЕК приводит теоретически к блокированию части каскада ведущего к ЕRK [64, 81, 79], но это является предметом дальнейших научных разработок.
В завершение, говоря еще раз о мультимодальном подходе с целью повышения эффективности и преодоления резистентности, следует рассматривать использование МЕК- ингибиторов как в комбинации с таргетами других сигнальных каскадов, например, фосфатидил-инозитол-3 киназы [78], так и комбинации с классическими цитостатиками. Онкологи с интересом ожидают результаты этих исследований, хотя во всех этих случаях, на сегодня их токсичность по-прежнему составляет определенную проблему (например с ингибитором АКТ или РI3К). Вместе с тем сейчас однозначно понятно, что со временем роль ингибиторов МЕК все более возрастает и рано или поздно они могут иметь большое значение в лечении злокачественных опухолей, особенно при использовании комбинированной терапии.
Библиографическая ссылка
Владимирова Л.Ю. ПРИМЕНЕНИЕ ИНГИБИТОРОВ МЕК В ОНКОЛОГИИ: РЕЗУЛЬТАТЫ И ПЕРСПЕКТИВЫ // Успехи современного естествознания. 2015. № 3. С. 18-30;URL: https://natural-sciences.ru/en/article/view?id=34730 (дата обращения: 11.07.2026).