


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
THE INTERACTION OF [4-OXOQUINAZOLIN-3(4H)-YL]ACETIC ACID ESTERS WITH GUANIDINE AND IT’S DERIVATIVES

Возросший в последние годы интерес к соединениям хиназолиновой природы обусловлен высокой фармакологической активностью различных функциональных производных этих гетероциклических соединений. В частности, в ряду N3-замещенных производных хиназолин-4(3Н)-она недавно обнаружены вещества, обладающие выраженными антидепрессантными [2], ноотропными [3], иммунотропными [4] свойствами, высокой противосудорожной [5] и антибактериальной [6] активностью. Относительная простота синтеза производных хиназолин-4(3Н)-она и широкий спектр их фармакологической активности позволяет отнести эти вещества к разряду «привилегированных молекул», на основе которых целесообразен дальнейший направленный поиск новых лекарственных кандидатов.
Цель исследования – cинтез новых функциональных производных хиназолин-4(3Н)-она на основе реакций нуклеофильного замещения в ряду сложных эфиров 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты при взаимодействии с гуанидином и его производными.
Материалы и методы исследования
Спектры ЯМР 1Н и 13С регистрировали на спектрометре «Bruker Avance 400» (400 МГц для 1H и 100 МГц для 13С) в ДМСО-D6, внутренний стандарт тетраметилсилан. Температуры плавления измерены в стеклянных капиллярах на приборе «Mel-Temp 3.0» (Laboratory Devices Inc., США). Синтез исходных сложных эфиров 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты был осуществлен путем региоселективного N3-алкилирования хиназолин-4(3Н)-она эфирами бромуксусной кислоты в среде безводного диметилформамида при температуре 100–105 °С в присутствии избытка карбоната калия, как это описано нами для α-галогенметилкетонов [1].
Изопропиловый эфир 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (I). 5,0 г (34,2 Ммоль) хиназолин-4(3Н)-она и 7,0 г (50,7 ммоль) свежепрокаленного и тонко измельченного карбоната калия перемешивают в 50 мл безводного диметилформамида при температуре 100–105 °С в течение 30 мин, добавляют в один прием раствор 6,5 г (35,9 ммоль) изопропилового эфира бромуксусной кислоты в 10 мл диметилформамида и перемешивают при той же температуре 1 ч. Охлаждают, фильтруют, фильтрат упаривают на роторном испарителе в вакууме при остаточном давлении 4–5 мм рт.ст. при температуре бани 80–85 °С, охлаждают, полученный остаток в виде вязкого масла растирают с 50 мл воды, выдерживают при температуре 0–5 °С в течение суток, образовавшийся осадок отфильтровывают, промывают 20 мл воды, сушат на воздухе, перекристаллизовывают из 25 мл изопропилового спирта и получают 7,3 г (87 %) светло-желтого кристаллического вещества, Тпл = 74–77 °С.
Спектр ЯМР 1Н, δ, м. д.: 1,23 д (6Н, 8 Гц, СН3); 4,65 c (2H, CH2); 7,45 т (1Н, 8 Гц, Н6); 7,68 т (1Н, 8 Гц, Н7); 7,71 д (1Н, 8 Гц, Н8); 7,96 с (1Н, Н2); 8,24 д (1Н, 8 Гц, Н5).
Спектр ЯМР 13С, δ, м. д.: 21,54; 47,35; 69,97; 121,66; 126,57; 127,28; 127,40; 134,33; 146,19; 147,90; 160,72; 166,79.
Бензиловый эфир 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (II) получают аналогично, выход 82 %, Тпл = 116–117 °С.
Спектр ЯМР 1Н, δ, м. д.: 4,93 с (2Н, OСН2); 5,22 c (2H, NCH2); 7,30–7,38 м (5Н, фенил); 7,57 т (1Н, 8 Гц, Н6); 7,71 д (1Н, 8 Гц, Н8) 7,85 т (1Н, 8 Гц, Н7); 7,98 с (1Н, Н2); 8,22 д (1Н, 8 Гц, Н5).
Спектр ЯМР 13С, δ, м. д.: 47,63; 66,93; 121,57; 126,35; 127,65; 127,70; 128,29; 128,56; 128,79; 135,04; 135,78; 148,21; 160,52; 168,24.
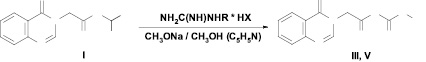
N-[2-[4-Оксохиназолин-3(4Н)-ил]ацетил]гуанидин (III). К раствору 5,0 г (20,3 ммоль) изопропилового эфира 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (I) в 50 мл метилового спирта добавляют 3,0 г (30,4 ммоль) гуанидина гидрохлорида, нагревают до кипения, добавляют в один прием свежеприготовленный раствор 0,75 г (32,6 мг-ат) натрия в 10 мл безводного метилового спирта и кипятят 15 мин. Горячий раствор фильтруют, отделяя образовавшийся натрия хлорид, фильтрат охлаждают, выдерживают при температуре 0–5 °С в течение суток, образовавшийся осадок отфильтровывают, промывают 10 мл холодного метилового спирта и 10 мл диэтилового эфира, сушат на воздухе и получают 4,4 г (87 %) белого кристаллического ватообразного вещества, Тпл = 240–242 °С.
Спектр ЯМР 1Н, δ, м. д.: 4,39 c (2H, CH2); 7,50 т (1Н, 8 Гц, Н6); 7,54 уш.с. (4Н, NH); 7,65 д (1Н, 8 Гц, Н8); 7,78 т (1Н, 8 Гц, Н7); 8,12 д (1Н, 8 Гц, Н5); 8,22 с (1Н, Н2).
Спектр ЯМР 13С, δ, м. д.: 49,50; 121,95; 126,32; 127,05; 127,27; 134,37; 148,31; 149,29; 158,95; 160,59; 171,65.
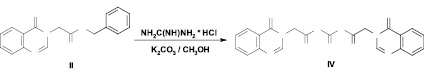
N,N’-Ди[2-[4-оксохиназолин-3(4Н)-ил]ацетил]гуанидин (IV). К раствору 2,0 г (8,1 ммоль) изопропилового эфира 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (I) в 50 мл метилового спирта добавляют 1,0 г (10,5 ммоль) гуанидина гидрохлорида и 3,0 г (21,7 ммоль) свежепрокаленного и тонко измельченного карбоната калия и кипятят в течение 8 ч. Горячий раствор фильтруют, фильтрат упаривают в вакууме досуха, твердый остаток экстрагируют 50 мл кипящего 95 % этилового спирта, охлаждают и выдерживают при температуре 0–5 °С в течение суток. Образовавшийся осадок отфильтровывают и получают 1,1 г (63 %) белого кристаллического вещества, Тпл = 309–312 °С.
Спектр ЯМР 1Н, δ, м. д.: 4,32 c (2H, CH2); 7,48 т (1Н, 8 Гц, Н6); 7,63 д (1Н, 8 Гц, Н8); 7,77 т (1Н, 8 Гц, Н7); 8,13 д (1Н, 8 Гц, Н5); 8,15 с (1Н, Н2).
Спектр ЯМР 13С, δ, м. д.: 49,60; 122,12; 126,33; 126,82; 127,28; 134,16; 148,44; 149,60; 159,65; 160,48; 169,81.
N-[2-[4-Оксохиназолин-3(4Н)-ил]ацетил]-N’-карбамоилгуанидин (V). К раствору 3,0 г (10,2 ммоль) бензилового эфира 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (II) в 25 мл безводного пиридина добавляют 1,75 г (11,6 ммоль) дициандиамидина сульфата и свежеприготовленный раствор 0,3 г (13,0 мг-ат) натрия в 10 мл безводного метилового спирта. Реакционную массу кипятят при интенсивном перемешивании в течение 2 ч и оставляют на ночь при комнатной температуре. Образовавшийся осадок отфильтровывают, промывают в 3 приема 15 мл изопропилового спирта, сушат на воздухе и получают 3,5 г смеси твердых продуктов реакции. Их экстрагируют 2 раза по 50 мл кипящего изопропилового спирта, объединенный экстракт охлаждают, образовавшийся осадок отфильтровывают и получают 1,35 г (46 %) светло-желтого кристаллического вещества, Тпл = 168,5–171,5 °С (разл.).
Спектр ЯМР 1Н, δ, м. д.: 4,45 с (2Н, СН2); 6,93 уш. с (2Н, NH); 7,52 т (1Н, 8 Гц, Н6); 7,66 д (1Н, 8 Гц, Н8); 7,80 т (1Н, 8 Гц, Н7); 8,14 д (1Н, 8 Гц, Н5); 8,28 с (1Н, Н2); 8,53 уш. с. (3Н, NH).
Спектр ЯМР 13С, δ, м. д.: 49,25; 122,02; 126,33; 127,03; 127,39; 134,35; 148,44; 149,27; 156,04; 156,79; 160,50; 172,59.
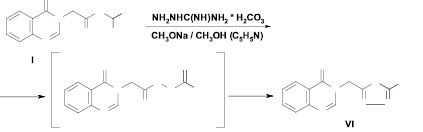
3-[(5-Амино-4Н-1,2,4-триазол-3-ил)метил]хиназолин-4(3Н)-он (VI). К раствору 3,0 г (12,2 ммоль) изопропилового эфира 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (I) в 25 мл безводного пиридина добавляют при интенсивном перемешивании 2,0 г (14,7 ммоль) аминогуанидина гидрокарбоната и свежеприготовленный раствор 0,4 г (17,4 мг-ат) натрия в 10 мл безводного метилового спирта. Реакционную массу кипятят при интенсивном перемешивании в течение 2 ч и оставляют на ночь при комнатной температуре. Образовавшийся осадок отфильтровывают, промывают в 3 приема 25 мл изопропилового спирта, сушат на воздухе и получают 4,0 г смеси твердых продуктов реакции. Их экстрагируют 4 раза по 50 мл кипящего 95 % этилового спирта, объединенный экстракт охлаждают, образовавшийся осадок отфильтровывают и получают 1,5 г (51 %) светло-желтого игольчатого вещества, Тпл = 328–331 °С (разл.).
Спектр ЯМР 1Н, δ, м. д.: 4,30 с (2Н, СН2); 7,50 т (1Н, 8 Гц, Н6); 7,64 д (1Н, 8 Гц, Н8); 7,80 т (1Н, 8 Гц, Н7); 8,13 д (1Н, 8 Гц, Н5); 8,27 с (1Н, Н2).
Спектр ЯМР 13С, δ, м. д.: 49,56; 120,07; 122,16; 126,32; 126,78; 127,27; 134,11; 148,47; 149,64; 160,44; 169,66.
Результаты исследования и их обсуждение
Взаимодействие сложных эфиров 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты с гуанидином протекает, в зависимости от условий реакции, с образованием N-моно- или N,N’-дизамещенного гуанидина. При осуществлении реакции в кипящем метиловом спирте в присутствии метилата натрия обеспечивается высокая концентрация гуанидина-основания в растворе, и основным продуктом реакции является N-[2-[4-оксохиназолин-3(4Н)-ил]ацетил]гуанидин (III) (R = H, X = Cl).
При использовании карбоната калия, практически не растворимого в метиловом спирте, реакция протекает значительно медленнее, и первоначально образующийся N-монозамещенный гуанидин (III) взаимодействует со второй молекулой сложного эфира, приводя к симметричному N,N’-ди[2-[4-оксохиназолин-3(4Н)-ил]ацетил]гуанидину (IV).
Попытка осуществления взаимодействия сложных эфиров I и II с аминогуанидина гидрокарбонатом и дициандиамидина сульфатом (R = C(O)NH2, X = HSO4) в кипящем метиловом спирте в присутствии метилата натрия не увенчалась успехом ввиду их очень низкой, в отличие от гуанидина гидрохлорида, растворимостью. Однако замена метилового спирта на безводный пиридин позволила получить желаемые продукты аминирования V и VI с выходом 46 и 51 %. При этом взаимодействие сложных эфиров 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты с аминогуанидином сопровождается дальнейшей циклизацией с образованием производного 1,3,4-триазола VI, строение которого дополнительно подтверждено масс-спектрометрией.
В масс-спектрах линейных продуктов аминирования III-V отсутствуют пики молекулярных ионов из-за их низкой стабильности и быстрого распада с отщеплением фрагментов цианамида (в случае III и IV) или цианмочевины (в случае V) и образованием одинакового во всех случаях катион-радикала амида 2-[4-оксохиназолин-3(4Н)-ил]уксусной кислоты (m/z 204), далее распадающегося до 3-метилхиназолин-4(3Н)-она (m/z 160). В противоположность этому продукт циклизации VI обнаруживает в своем масс-спектре достаточно интенсивный пик более стабильного молекулярного иона (m/z 242).
Дополнительно наличие в структуре соединения III монозамещенного гуанидинового фрагмента было доказано реакцией Сакагучи по образованию красного окрашивания раствора при окислении гипохлоритом натрия в присутствии α-нафтола.
Фармакологические свойства новых соединений III–VI изучаются.
Заключение
В результате проведенных исследований осуществлен синтез новых производных хиназолин-4(3Н)-она, содержащих линейные или циклические фрагменты гуанидина в своей структуре.
Библиографическая ссылка
Озеров А.А., Новиков М.С., Глухова Е.Г. ВЗАИМОДЕЙСТВИЕ СЛОЖНЫХ ЭФИРОВ [4-ОКСОХИНАЗОЛИН-3(4Н)-ИЛ]-УКСУСНОЙ КИСЛОТЫ С ГУАНИДИНОМ И ЕГО ПРОИЗВОДНЫМИ // Успехи современного естествознания. 2016. № 2. С. 53-56;URL: https://natural-sciences.ru/en/article/view?id=35787 (дата обращения: 17.07.2026).