


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
SYNTHESIS OF ACYL DERIVATIVES N-SUBSTITUTED AROMATIC CARBAMATES AND THEIR SOME TRANSFORMATIONS

Перхлораты металлов широко используются в качестве кислот Льюиса, катализирующих разнообразные превращения органических веществ, в том числе реакции этерификации, защиты и снятия защитных групп, присоединение по Михаэлю, присоединение с раскрытием цикла, конденсации, a-галогенирование 1,3-дикарбонильных соединений, синтез трет-бутиловых простых эфиров, этилкарбонатов и других соединений [4]. Они имеют заметную способность хелатировать ряд соединений, например, 1,3-дикарбонильных систем [7], а также характеризуются уникальными свойствами, среди которых следует отметить значительный ионный характер, электроотрицательность, которая соответствует высокой энергии сольватации, что обуславливает их способность хорошо растворяться в воде и в большом числе неводных растворителей [13].
В обзоре [8], посвященном применению в органической химии хлорной кислоты и ее солей, рассмотрены реакции, в которых проявляется каталитическое действие этих соединений. Важнейшей среди таких реакций является реакция ацилирования ароматических и гетероароматических ядер по Фриделю – Крафтсу.
Ацильные производные аренов и гетаренов широко используются в органическом синтезе, в частности для получения халконов и различных гетероциклических систем. Из перхлоратов в реакции ацилирования наиболее широко применяются перхлораты лития, магния, цинка и никеля [4]. Показано, что при обработке активированного электронодонорными заместителями (ОМе, Ме) бензола эквимолярным количеством ангидрида в присутствии двух эквивалентов перхлората лития или 0,5-1 мол % Mg(ClO4)2 при 60 °С с высоким выходом образуются соответствующие продукты ацилирования. Методом ЯМР 1Н спектроскопии показано, что перхлорат металла сильно координируется с ангидридом, ускоряя, таким образом, электрофильную атаку по бензольному кольцу [5].
Продукты ацилирования фенолов, тиолов, аминов и спиртов являются важными полупродуктами в синтезе лекарственных и фармацевтических препаратов, которые обычно получают по реакции ацилирования ангидридами [9]. В то же время слабые нуклеофильные свойства НО-содержащих соединений, в особенности фенолов, требуют активации ангидрида, которая обычно достигается применением таких нуклеофильных агентов, как DMAP [12], Bu3P [14] и кислот Льюиса СoCl2 [2], трифлатов скандия, висмута и индия [4], цеолитов [3], глин [10], HBF4 – SiO2 [6]. Однако эти методы ацилирования не лишены недостатков с позиций принципов зеленой химии. К их числу относится значительная продолжительность реакций, жесткие условия, применение галогенсодержащих растворителей, высокотоксичных веществ (DMAP) или легковоспламеняющихся (Bu3P) веществ, дорогостоящих трифлатов, необходимость применения избытка ацилирующего агента, а также возможность протекания побочных процессов в случае субстратов, чувствительных в кислой среде.
Хорошо известно, что некоторые реакционноспособные соединения, например электроноизбыточные арены и гетероциклические соединения (тиофен, сильван), способны подвергаться С-ацилированию при помощи системы реагентов уксусный ангидрид – перхлорат магния [1]. Так, авторы статьи [11] показали, что 5-фенил-2-(фур-2-ил)оксазол подвергается ацилированию системой Ас2О – Mg(ClO4)2 с образованием 5-фенил-2-(5-ацетилфур-2-ил)оксазола с выходом 60 %.
Реакции С- и О-ацилирования ароматических N-замещенных карбаматов и их некоторые превращения
Нами установлено, что ацилирование метил N-3(4)[(метоксикарбонил)амино]фенилкарбаматов (I, II) эквимолярным количеством уксусного ангидрида в присутствии 1 мол % перхлората магния при нагревании реакционной массы при 70 °С в течение 1 ч протекает с образованием соответствующих ацильных производных (III, IV) с выходами 78–85 %.
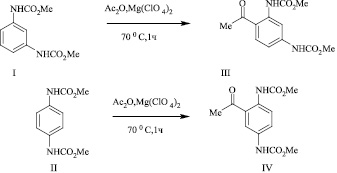
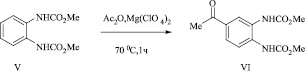
В случае метил N-2-[(метоксикарбонил)амино]фенилкарбамата (V) реакция С-ацилирования в аналогичных условиях протекает селективно с образованием одного из возможных региоизомеров (VI), структура которого подтверждена методом ЯМР 1Н спектроскопии.
Роль перхлоратов металлов в активации ангидрида заключается в координации катиона перхлората (MLn) ангидридом, что приводит к образованию шестичленного переходного состояния А [7].
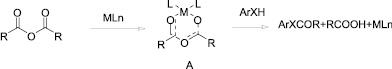
В переходном состоянии А ангидрид становится более восприимчивым к нуклеофильной атаке ареном, которая ведет к образованию продукта ацилирования, карбоновой кислоты и регенерации катализатора.
При попытке ацилировать этой системой реагентов метил N-(4-гидроксифенил)карбамата (VII) при комнатной температуре и при перемешивании в течение 20 мин, а также при использовании в качестве катализатора концентрированной серной кислоты при нагревании при 80–90 °С, в обоих случаях был выделен только продукт О-ацилирования (VIII) с выходами 85 и 79 % соответственно.
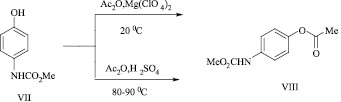
Авторы статьи [7] показали, что большее соотношение величины заряда к размеру иона Mg2+ по сравнению с ионом Li+ дает возможность перхлорату магния координироваться с ангидридом более эффективно и в более мягких условиях, поэтому с достаточно активированными соединениями, к числу которых принадлежат производные фенолов, реакцию проводят при комнатной температуре.
Структура 4-[(метоксикарбонил)амино]фенилацетата (VIII) подтверждена методом ИК спектроскопии и элементным анализом.
В ИК спектре О-ацильного производного (VIII) в отличие от исходного гидроксизамещенного карбамата (VII) отсутствует полоса поглощения при 3220 см–1, обусловленная валентными колебаниями фенольного гидроксила, но в то же время присутствует полоса поглощения при 3335 см–1, связанная с валентными колебаниями группы NH, а также наряду с полосами валентных колебаний связей  бензольного кольца имеются полосы поглощения при 1710 и 1780 см–1, обусловленные колебанием карбонилов карбаматной и ацетоксигрупп.
бензольного кольца имеются полосы поглощения при 1710 и 1780 см–1, обусловленные колебанием карбонилов карбаматной и ацетоксигрупп.
Нами изучена возможность перегруппировки О-ацильного производного в продукт С-ацилирования. Установлено, что при нагревании 4-[(метоксикарбонил)амино]фенилацетата (VIII) в течение 3 ч в дихлорэтане в присутствии безводного хлорида алюминия образуется метил N-(3-ацетил-4-гидроксифенил)карбамат (IX) с выходом 64 %.
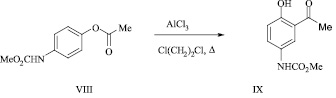
Строение соединения (IX) подтверждено методами ИК и ЯМР 1Н спектроскопии.
С целью дальнейшей функционализации соединения IX альдольно-кротоновой конденсацией Кляйзена –Шмидта с вератровым альдегидом в условиях основного катализа получен халкон (X).
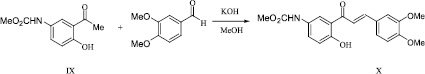
Структура метил N-{3-[(E)-3-(3,4-диметоксифенил)-2-пропеноил]-4-гидроксифенил}карбамата (X) подтверждена методами ИК и ЯМР 1Н спектроскопии.
В спектре ЯМР 1Н протоны сопряженной двойной связи проявляются в области 7,19–7,24 м.д. (J 15,2 Гц), что свидетельствует о Е-конфигурации халкона (X).
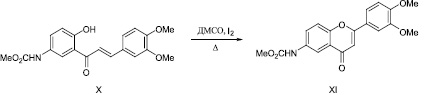
Установлено, что кипячение соединения (X) в ДМСО в присутствии каталитического количества иода сопровождается гетероциклизацией с образованием метил N-[2-(3,4-диметоксифенил)-4-оксо-3,4-дигидро-2Н-хромен-6-ил]карбамата (XI).
Экспериментальная часть
Спектры ЯМР 1Н получены на спектрометре Bruker DRX-500 (500,13 МГц), растворитель ДМСО-d6. ИК спектры измерены на ИК Фурье-спектрофотометре InfraLUM FT-02 в интервале 4000–400 см–1 в KBr. Масс-спектры получены на приборах МХ-1306; Kratos MS-30 и Finigan MAT INCOS 50 при энергии ионизирующих электронов 70 эВ. Чистоту полученных соединений контролировали методом ТСХ на пластинах Silufol UV-254; проявление в парах иода.
Метил N-{2-ацетил-5-[(метоксикарбонил)амино]фенил}карбамат (III). Смесь 2,24 г (0,01 моль) метил N-3-[(метоксикарбонил)амино]фенилкарбамата (I), 1,2 мл (0,012 моль) уксусного ангидрида и 22 мг (1 мол %) перхлората магния нагревали на водяной бане при 70 °С в течение 1 ч, охлаждали до комнатной температуры, добавляли 10 мл ледяной воды, осторожно нейтрализовали кислоту твердым карбонатом натрия и полученную смесь обрабатывали бензолом (2×15 мл). Объединенные бензольные экстракты сушили сульфатом магния и растворитель удаляли в вакууме, полученный кристаллический продукт подвергали хроматографированию на нисходящей стеклянной колонке, заполненной активированным силикагелем марки Silicagel 100/400 мкм, элюент – метиленхлорид – диэтиловый эфир, 1:1 (по объему) и перекристаллизовывали из диэтилового эфира. Выход 2,3 г (87 %), бесцветные кристаллы, Тпл = 124–126 °С. ИК спектр, n, см–1: 3310; 3330 (NH), 1710; 1680 (С=O), 1610; 1585; 1565 (С=С, 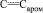 .). Спектр ЯМР 1Н, d, м.д.: 2,58 с (3Н, СОMe), 3,70 с (6Н, 2NHCO2Me), 7,28 д (1 Наром, J 8 Гц), 7,99 д (1 Наром, J 8 Гц), 8,53 с (1 Наром), 10,18 уш. с (1Н, NH), 11,39 уш. с (1 Н, NH). Масс-спектр, m/z (Iотн, %): 267 (8) [M+1]+, 266 (52) [M]+, 251 (13), 235 (5), 224 (13), 220 (10), 219 (100), 192 (5), 187 (28), 175 (5), 147 (6), 133 (4), 119 (2), 78 (2), 59 (7), 43 (5). Найдено, %: С 53,81; Н 5,25; N 10,42. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53. M 266.
.). Спектр ЯМР 1Н, d, м.д.: 2,58 с (3Н, СОMe), 3,70 с (6Н, 2NHCO2Me), 7,28 д (1 Наром, J 8 Гц), 7,99 д (1 Наром, J 8 Гц), 8,53 с (1 Наром), 10,18 уш. с (1Н, NH), 11,39 уш. с (1 Н, NH). Масс-спектр, m/z (Iотн, %): 267 (8) [M+1]+, 266 (52) [M]+, 251 (13), 235 (5), 224 (13), 220 (10), 219 (100), 192 (5), 187 (28), 175 (5), 147 (6), 133 (4), 119 (2), 78 (2), 59 (7), 43 (5). Найдено, %: С 53,81; Н 5,25; N 10,42. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53. M 266.
Соединения (IV, VI) получены аналогично.
Метил N-{2-ацетил-4-[(метоксикарбонил)амино]фенил}карбамат (IV). Выход 2,35 г (89 %), бесцветные кристаллы, Tпл = 119-120 °С (из этанола). ИК спектр, n, см–1: 3315; 3330 (NH), 1710; 1675 (С=O), 1610; 1575; 1565 (С=С, 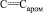 .). Спектр ЯМР 1Н, d, м.д.: 2,53 c (3 H, COMe), 3,70 с (6Н, 2NHCO2Me), 7,08 д (1 Наром, J 8 Гц), 7,22 с (1 Наром), 7,48 д (1 Наром, J 8 Гц), 9,49 уш. с (1 Н, NH), 9,75 уш. с (1 Н, NH). Масс-спектр, m/z (Iотн, %): 267 (2) [M+1]+, 266 (16) [M]+, 235 (2), 225 (14), 224 (100), 208 (5), 193 (11), 192 (67), 176 (2) 165 (13), 160 (31), 147 (8), 133 (13), 121 (11), 106 (8), 94 (6), 78 (6), 59 (19), 43 (33). Найдено, %: С 54,01; Н 5,21; N 10,35. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53. M 266.
.). Спектр ЯМР 1Н, d, м.д.: 2,53 c (3 H, COMe), 3,70 с (6Н, 2NHCO2Me), 7,08 д (1 Наром, J 8 Гц), 7,22 с (1 Наром), 7,48 д (1 Наром, J 8 Гц), 9,49 уш. с (1 Н, NH), 9,75 уш. с (1 Н, NH). Масс-спектр, m/z (Iотн, %): 267 (2) [M+1]+, 266 (16) [M]+, 235 (2), 225 (14), 224 (100), 208 (5), 193 (11), 192 (67), 176 (2) 165 (13), 160 (31), 147 (8), 133 (13), 121 (11), 106 (8), 94 (6), 78 (6), 59 (19), 43 (33). Найдено, %: С 54,01; Н 5,21; N 10,35. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53. M 266.
Метил N-{4-ацетил-2-[(метоксикарбонил)амино]фенил}карбамат (VI). Выход 2,29 г (87 %), бесцветные кристаллы, Тпл = 138140 °С (из метанола). ИК спектр, n, см–1: 3310; 3325 (NH), 1710; 1685 (С=O), 1610; 1570; 1562 (С=С, 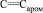 .). Спектр ЯМР 1Н, d, м.д.: 2,54 c (3 H, COMe), 3,76 с (6 Н, 2NHCO2Me), 7,84 д (1 Наром, J 8,5 Гц), 7,94 с (1 Наром), 7,98 д (1 Наром, J 8,5 Гц), 9,54 уш. с (1 Н, NH), 9,80 уш. с (1 Н, NH). Найдено, %: С 54,09; Н 5,20; N 10,48. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53.
.). Спектр ЯМР 1Н, d, м.д.: 2,54 c (3 H, COMe), 3,76 с (6 Н, 2NHCO2Me), 7,84 д (1 Наром, J 8,5 Гц), 7,94 с (1 Наром), 7,98 д (1 Наром, J 8,5 Гц), 9,54 уш. с (1 Н, NH), 9,80 уш. с (1 Н, NH). Найдено, %: С 54,09; Н 5,20; N 10,48. C12H14N2O5. Вычислено, %: С 54,14; Н 5,26; N 10,53.
4-[(Метоксикарбонил)амино]фенилацетат (VIII).
а) Смесь 1,67 г (0,01 моль) метил-N-(n-гидроксифенил)карбамата (VII), 1,2 мл (0,012 моль) свежеперегнанного уксусного ангидрида, 2 капли концентрированной серной кислоты нагревали на кипящей водяной бане в течение четырех часов, реакционную массу охлаждали и выливали на 50 г толченого льда. Выпавший кристаллический осадок отфильтровывали, промывали водой, сушили на воздухе и перекристаллизовывали из гексана. Выход 1,65 г (79 %) соединения (VIII), бесцветные кристаллы, Тпл = 77–79 °С. ИК спектр, n, см–1: 3335 (NH), 1710; 1780 (С=О), 1620; 1555; 1530 (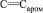 ). Найдено, %: С 57,42; Н 5,26; N 6,62. C10H11NO4. Вычислено, %: С 57,28; Н 5,30; N 6,39.
). Найдено, %: С 57,42; Н 5,26; N 6,62. C10H11NO4. Вычислено, %: С 57,28; Н 5,30; N 6,39.
б) Смесь 1,67 г (0,01 моль) метил N (4-гидроксифенил)карбамата (VII), 1,2 мл (0,012 моль) уксусного ангидрида и 22 мг (1 мол %) перхлората магния перемешивали при 20 °С в течение 20 мин, выливали на измельченный лед, выпавший осадок отфильтровывали, сушили на воздухе и перекристаллизовывали из гексана. Выход 1,88 г (90 %), бесцветные кристаллы, Тпл = 77–79 °С. Найдено, %: С 57,31; Н 5,28; N 6,35. C10H11NO4. Вычислено, %: С 57,28; Н 5,30; N 6,39.
Метил N-(3-ацетил-4-гидроксифенил)карбамат (IX). Смесь 1 г (5 ммоль) 4-[(метоксикарбонил)амино]фенилацетата (VIII), 0,5 г безводного хлорида алюминия в 15 мл дихлорэтана кипятили 5 ч, охлаждали, обрабатывали 1 н раствором соляной кислоты (50 мл), органический слой промывали водой, сушили сульфатом магния и растворитель удаляли, остаток перекристаллизовывали из гексана. Выход 0,89 г (89 %), бесцветные кристаллы, Тпл = 98-99 °С. ИК спектр, n, см–1: 3315; 3400 (NH,ОН), 1710; 1690 (С=О), 1610; 1565; 1550 (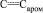 ). Спектр ЯМР 1Н, d, м.д.: 2,75 с (3 Н, СОМе), 3,71 с (3 Н, NHCO2Me), 7,04 д (1 Н, Наром, J 8,5 Гц), 7,54 д (1 Н, Наром, J 8,5 Гц), 7,95 с (1 Н, Наром.), 9,58 уш. с (1 Н, NH), 13,14 c (1 Н, OH). Найдено, %: С 57,31; Н 5,30; N 6,43. C10H11NO4. Вычислено, %: С 57,28; Н 5,30; N 6,39.
). Спектр ЯМР 1Н, d, м.д.: 2,75 с (3 Н, СОМе), 3,71 с (3 Н, NHCO2Me), 7,04 д (1 Н, Наром, J 8,5 Гц), 7,54 д (1 Н, Наром, J 8,5 Гц), 7,95 с (1 Н, Наром.), 9,58 уш. с (1 Н, NH), 13,14 c (1 Н, OH). Найдено, %: С 57,31; Н 5,30; N 6,43. C10H11NO4. Вычислено, %: С 57,28; Н 5,30; N 6,39.
Метил N-{3-[(E)-3-(3,4-диметоксифенил)-2-пропеноил]-4-гидроксифенил}карбамат (X). К смеси 0,42 г (2 ммоль) метил N-(3-ацетил-4-гидроксифенил)карбамата (IX), 0,33 г (2 ммоль) 3,4-диметоксибензальдегида в 10 мл метанола при 35 °С в течение 0,5 ч при перемешивании добавляли 0,6 мл 10 %-го метанольного раствора гидроксида калия. Реакционную массу перемешивали 4 ч при 35 °С, оставляли на 24 ч при комнатной температуре, выливали в 100 мл ледяной воды, подкисляли разбавленной соляной кислотой (1:1). Выпавший осадок отфильтровывали, сушили на воздухе и перекристаллизовывали из этанола. Выход 0,76 г (84 %), светло-желтые кристаллы, Тпл = 162–164 °С. ИК спектр, n, см–1: 3320; 3405 (NH, OH), 1710; 1680 (С=O), 1610; 1585; 1565 (С=С, 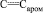 ). Спектр ЯМР 1Н, d, м.д.: 3,71 с (3 Н, NHCO2Me), 3,85 c (6 H, 2OMe), 6,79 д (1 Наром, J 8,2 Гц), 7,02 c (1 Наром), 7,15 д (1 H, Hаром, J 8,2 Гц), 7,19–7,21 м (2 Н, 1 Наром, 1 Н, НС=СН), 7,24 д (1 Н, НС=СН, J 15,2 Гц), 7,67 д (1 Наром, J 8,5 Гц), 8,15 с (1 Наром), 9,59 уш. с (1 Н, NH), 13,14 с (1 Н, ОН). Найдено, %: С 63,84; Н 5,34; N 4,01. C19H19NO6. Вычислено, %: С 63,87; Н 5,32; N 3,92.
). Спектр ЯМР 1Н, d, м.д.: 3,71 с (3 Н, NHCO2Me), 3,85 c (6 H, 2OMe), 6,79 д (1 Наром, J 8,2 Гц), 7,02 c (1 Наром), 7,15 д (1 H, Hаром, J 8,2 Гц), 7,19–7,21 м (2 Н, 1 Наром, 1 Н, НС=СН), 7,24 д (1 Н, НС=СН, J 15,2 Гц), 7,67 д (1 Наром, J 8,5 Гц), 8,15 с (1 Наром), 9,59 уш. с (1 Н, NH), 13,14 с (1 Н, ОН). Найдено, %: С 63,84; Н 5,34; N 4,01. C19H19NO6. Вычислено, %: С 63,87; Н 5,32; N 3,92.
Метил N-[2-(3,4-диметоксифенил)-4-оксо-4Н-хромен-6-ил]карбамат (XI). Растворяли 0,1 г (0,4 ммоль) иода в 10 мл диметилсульфоксида, добавляли 0,71 г (2 ммоль) халкона (X), полученную смесь кипятили 0,5 ч, охлаждали, переносили на измельченный лед, добавляли 10 %-ный раствор тиосульфата натрия для удаления иода, осадок отфильтровывали, сушили на воздухе и перекристаллизовывали из этанола. Выход 0,55 г (78 %), бесцветные кристаллы, Тпл = 201-203 °С. ИК спектр, n, см–1: 3310 (NH), 1710; 1655 (С=O), 1610; 1575; 1560 (С=С, 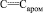 ). Спектр ЯМР 1Н, d, м.д.: 3,71 с (3 Н, NHCO2Me), 3,80 с (6 Н, 2ОМе), 6,64 с (1 Нхромена), 6,85 д (1 Н, Наром, J 7,5 Гц), 7,15 с (1 Н, Наром), 7,48–7,51 м (2 Н, Наром), 7,82 д (1 Н, Наром., J 8,5 Гц), 8,25 с (1 Н, Наром), 9,58 уш. с (1 Н, NH). Найдено, %: С 63,99; Н 4,58; N 4,01. C19H17NO6. Вычислено, %: С 64,23; Н 4,79; N 3,94.
). Спектр ЯМР 1Н, d, м.д.: 3,71 с (3 Н, NHCO2Me), 3,80 с (6 Н, 2ОМе), 6,64 с (1 Нхромена), 6,85 д (1 Н, Наром, J 7,5 Гц), 7,15 с (1 Н, Наром), 7,48–7,51 м (2 Н, Наром), 7,82 д (1 Н, Наром., J 8,5 Гц), 8,25 с (1 Н, Наром), 9,58 уш. с (1 Н, NH). Найдено, %: С 63,99; Н 4,58; N 4,01. C19H17NO6. Вычислено, %: С 64,23; Н 4,79; N 3,94.
Заключение
Приведенные в работе экспериментальные данные показывают эффективность и региоселективность реакций ацилирования изомерных ароматических дикарбаматов и 4-гидроксизамещенного фенилкарбамата уксусным ангидридом в присутствии каталитических количеств перхлората магния, а также синтетические возможности ацилпроизводных при получении некоторых новых производных арил- и гетарилкарбаматов.
Библиографическая ссылка
Великородов А.В., Шустова Е.А., Степкина Н.Н., Старикова А.А. СИНТЕЗ АЦИЛПРОИЗВОДНЫХ N-ЗАМЕЩЕННЫХ АРОМАТИЧЕСКИХ КАРБАМАТОВ И ИХ НЕКОТОРЫЕ ПРЕВРАЩЕНИЯ // Успехи современного естествознания. 2016. № 7. С. 21-26;URL: https://natural-sciences.ru/en/article/view?id=35996 (дата обращения: 23.06.2026).
DOI: https://doi.org/10.17513/use.35996