


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
THE PERSPECTIVE METHOD OF SYNTHESIS OF DIMETHYLDIETHYLBUTYNDIOL

Ацетилен – один из крупнейших полупродуктов промышленного органического синтеза. Его мировое производство превышает 2 млн тонн в год. Благодаря наличию тройной связи и подвижных атомов водорода ацетилен обладает высокой реакционной способностью, которая и определяет его применение для производства многих продуктов основного и тонкого органического синтеза [1]. Широкое использование ацетилена сдерживается в настоящее время его высокой стоимостью по сравнению с этиленом. Однако устойчивое повышение цен на нефть в недалеком будущем позволит ацетилену стать конкурентоспособным сырьем для химической промышленности по отношению к пока более дешевому этилену. Особый интерес промышленные ацетиленовые технологии органического синтеза вызывают в первую очередь в тех странах, которые богаты углем (Германии, Англии и некоторых других). В них на основе карбидного ацетилена сформировалась большая ветвь промышленного органического синтеза.
Производные ацетилена, представляющие его соединения, в которых один или оба атома водорода заменены на какой-либо другой атом или группу атомов, имеют всё большую перспективу применения для целей тонкого органического синтеза. На основе ацетиленовых соединений уже сейчас созданы разработки для производства лекарственных препаратов, специальных полимеров, фотоактивных веществ, жидких кристаллов и многих других соединений, используемых в новой технике. Огромный интерес представляют ацетиленовые спирты, особенно гликоли, так как наличие в их составе совместно с ацетиленовой группой, спиртовых групп и алкильных радикалов дает возможность получать разнообразные винильные мономеры и создавать полимеры на их основе [1]. Они являются ценными продуктами с широким спектром практического применения – полупродукты в синтезе душистых веществ, лекарственных препаратов и витаминов, антикоррозийные средства для защиты нефтяного оборудования, модификаторы моторных топлив, эмульгаторы и другие соединения, используемые в современной технике.
Целью данного исследования был синтез в лабораторных условиях на основе реакции Фаворского нового мономера диметилдиэтилбутиндиола (по номенклатуре IUPAC 3,6–диметил–3,6-дигидрокси–октин–4) со следующей структурной формулой [2]:
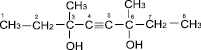
Данное соединение относится к органической химии, в частности к функциональным производным ацетилена в области полимеров, которое содержит в своей молекуле одновременно две гидроксильные группы и тройную связь, что дает возможность получать из него полимеры с заданными характеристиками, лекарства и витамины, душистые вещества, эмульгаторы и флотореагенты. Применение производных данного соединения в качестве флотореагента при обогащении медных и серебряных руд на данный момент является наиболее актуальным, ввиду того, что в республике Казахстан флотореагенты не производятся, а ввозятся из-за рубежа [3].
Материалы и методы исследования
Известно, что получение ацетиленовых спиртов основано на взаимодействии ацетилена с карбонильными соединениями (технологии А.Е. Фаворского и В. Реппе). Еще в 1905 г. А.Е. Фаворским была высказана мысль о возможности прямой конденсации карбонильных соединений с монозамещенными ацетиленами (алкинами с концевой тройной связью) в присутствии сильных оснований. И действительно, уже первое осуществленное в этом направлении взаимодействие – конденсация ацетона с фенилацетиленом в присутствии порошкообразного едкого калия – позволило одностадийно и с количественным выходом получить ожидаемый 2-метил-4-фенилбутан-3-ол-2 [1]. Впоследствии эта реакция была распространена не только на прочие монозамещенные ацетилены, но и на сам ацетилен.
Весьма обстоятельные исследования по определению оптимальных условий проведения данной реакции были выполнены И.А. Фаворской с сотрудниками. Было показано, что образованию гликолей в эфирной среде способствуют:
а) медленное перемешивание реакционной, массы;
б) слабый ток ацетилена;
в) быстрое добавление карбонильной компоненты.
Знание этих закономерностей позволило упомянутым авторам ввести в конденсацию не только такие малоактивные карбонильные соединения, как метил-н-нонилкетон, но и получить с количественными выходами ряд вторичных ацетиленовых карбинолов. Последний факт тем более значителен, что при осуществлении классического варианта реакции Фаворского взаимодействие ацетилена с алифатическими альдегидами очень часто сопровождается альдольной конденсацией взятого альдегида.
Синтез через ацетилениды щелочных металлов в среде инертных растворителей долгое время заметно сдерживался тем, что приходилось работать с грубодисперсными суспензиями этих агентов. Это осложняло температурный контроль реакций и очень часто приводило к разного рода неудачам при синтезе ацетиленовых спиртов. К настоящему времени разработан ряд легко воспроизводимых даже в лабораторных условиях прямых способов получения тонкодисперсных суспензий ацетиленидов щелочных металлов в инертных растворителях, что значительно расширило синтетические возможности указанного выше метода [4].
Ни один из методов стехиометрического этинилирования карбонильных соединений в среде инертных растворителей не используется в настоящее время столь широко, как этинилирование в среде жидкого аммиака. Удобства в проведении такого рода синтезов общеизвестны, поэтому имеет смысл лишь отметить, что интенсивная разработка этого способа началась после сообщения К. Кэмпбелла с сотрудниками о синтезе большого числа ацетиленовых спиртов с помощью ацетиленидов натрия в жидком аммиаке. На основании большого экспериментального материала было выяснено, что из всех применяемых ацетиленидов наиболее мягко действующим является ацетиленид лития, наиболее реакционноспособен – ацетиленид калия, а ацетиленид натрия занимает промежуточное между ними положение [1, 4].
Преимущество предлагаемой в данном исследовании технологии получения ацетиленовых спиртов перед уже известными заключается в безопасности, экономичности и более высокой интенсивности процесса: не применяется жидкий аммиак (токсичный, раздражающий растворитель, образующий с воздухом взрывоопасные смеси), ацетилен используется при атмосферном давлении, процесс проводится при температуре, близкой к комнатной, нет вредных выбросов и отходов. Получение ацетиленовых спиртов производится присоединением ацетиленовых углеводородов к карбонильным соединениям в присутствии оснований. В реакцию вступают алициклические кетоны и некоторые альдегиды. В ряду ацетиленов чаще используют незамещенный ацетилен и винилацетилен. Обычно реакцию проводят с суспензией KOH в растворителе (эфире, бензоле, ДМФА и других) и большом избытке ацетилена (вместо ацетилена можно использовать CaC2 в присутствии KOH) [5].
Наиболее близким структурным аналогом диметилдиэтилбутиндиола является тетраметилбутиндиол
(СН3)2˗С(ОН)˗С≡С˗С(ОН)˗(СН3)2,
который также используют в качестве полупродуктов в синтезе лаков, клеев, флотореагентов, ингибиторов коррозии, душистых и лекарственных веществ [3]. Его получают при взаимодействии ацетилена с ацетоном в присутствии мелкодисперсного гидроксида калия в среде бензола или диэтилового эфира:
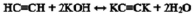
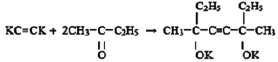
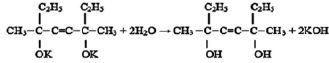
Недостатком данного вещества является короткая углеродная цепь, которая не позволяет получить большое количество производных. Поэтому в ходе данного исследования и стояла задача, используя реакцию Фаворского, синтезировать новое химическое соединение с более длинной углеродной цепью 3,6-диметил-3,6-дигидрокси-октин-4, путем присоединения ацетилена к бутанону-2 в присутствии порошкообразного КОН.
В результате данной реакции образуется соответствующий ацетиленид, при гидролизе которого с выходом 60 % и получают этот ацетиленовый спирт – диметилдиэтилбутиндиол.
Аппаратура для проведения синтеза состоит из трехгорлой колбы емкостью 0,5 л, снабженной механической мешалкой с ртутным затвором, обратным холодильником, капельной воронкой и трубкой для подачи ацетилена. Ацетилен для очистки пропускают через промывную склянку Тищенко с концентрированной H2SО4. Конец обратного холодильника также соединяется со склянкой Тищенко или счетчиком пузырьков, заполненными H2SO4, что защищает систему от попадания влаги и позволяет следить за поглощением ацетилена [5].
В трехгорлую колбу загружается 73 г порошка КОН, 270 мл бензола и 24,5 г бутанона-2. При перемешивании и охлаждении водопроводной водой (13–15 °С) через реакционную смесь в течение 1,5 часов пропускают 6 л очищенного ацетилена (из газометра). Реакционную смесь оставляют на ночь и затем гидролизуют, прибавляя из капельной воронки 70–80 мл дистиллированной воды. Бензольный слой отделяют в делительной воронке, а водный экстрагируют тремя порциями бензола по 30 мл. Бензольный слой и вытяжки от экстракции водного слоя соединяют, последовательно промывают 20 мл воды, 20 мл 20 %-ной уксусной кислоты, 20 мл воды и сушат прокаленным поташом. После отгонки бензола целевой продукт (3,6-диметил-3,6-дигидрокси-октин-4) кристаллизуется в колбе в виде игольчатых кристаллов желтоватого цвета с температурой плавления 59 °С, для очистки которых используют перекристаллизацию четыреххлористым углеродом.
Для идентификации и установления структуры полученного вещества были сняты 1Н и 13С ЯМР спектры на спектрометре JNM-ЕСА-400 компании «JEOL» (Япония) с рабочей частотой на ядрах водорода 400 МГц. Анализ выполняли одномерным методом ЯМР-спектроскопии, поскольку ядерный магнитный резонанс является сегодня одним из самых информативных методов исследования структуры и превращений молекул, межмолекулярных взаимодействий и количественного анализа веществ.
Результаты исследования и их обсуждение
Полученный образец продукта (предположительно диметилдиэтилбутиндиола) представлял собой аморфный порошок желтого цвета, хорошо растворимый в хлороформе, поэтому 1Н и 13С ЯМР-спектры образца снимали в дейтерированном хлороформе. Полученные реальные и модельные 1Н и 13С ЯМР-спектры данного образца приведены на рис. 1 и 2.
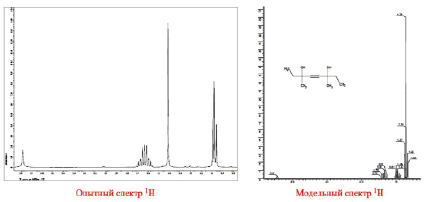
Рис. 1. Спектры 1Н ЯМР синтезированного образца диметилдиэтилбутиндиола
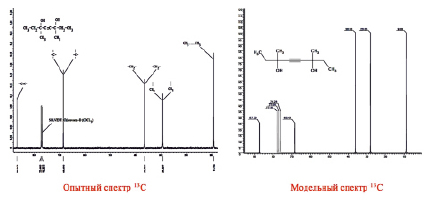
Рис. 2. Спектры 13С ЯМР синтезированного образца диметилдиэтилбутиндиола
Известно, что при исследовании методом ЯМР соединений с неизвестной структурой необходимо проводить также и изучение наиболее подходящих модельных соединений в идентичных условиях, т.к. это наиболее надежный путь её установления. Модельные спектры 1Н и 13С ЯМР предполагаемого соединения получали в программе ACDLab Chem Sketch Window, выполняющей большое количество всевозможных физико-химических расчетов.
Как видно из представленных на рис. 1 и 2 спектров, простота и симметричность анализируемого соединения однозначно проявилась в реальном и модельном вариантах. Это свидетельствует о правильной идентификации и установлении строения предполагаемого соединения.
Основываясь на наблюдениях за ходом проведенных экспериментов, можно также сказать, что предлагаемый метод синтеза ацетиленовых диолов отличается простотой реализации и легкостью выделения целевого продукта из реакционной смеси, имеет меньшую продолжительность (1,5 часа против 10 часов) и достаточно высокий выход целевого продукта (около 60 %). Кроме того, описанное вещество синтезируется на основе доступного химического сырья и в достаточно мягких условиях. Данные преимущества достигнуты благодаря учету рекомендованных [5–7] особенностей осуществления реакций данного типа: медленное перемешивание реакционной массы, слабый ток ацетилена и быстрое добавление кетона.
Выводы
Благодаря своей высокой ненасыщенности, ацетилен способен очень энергично присоединять самые разнообразные вещества. Возможности использования его в органическом синтезе настолько велики, что даже создана целая отрасль химической промышленности, в которой он является основным сырьевым источником.
Из всех непредельных углеводородов ацетилену принадлежит первое место по разнообразию продуктов, получаемых на его основе. Среди них находятся и ацетиленовые спирты, которые являются ценными продуктами с широким спектром практического применения. Они могут служить полупродуктами в синтезе душистых веществ, лекарственных препаратов и витаминов, антикоррозийными средствами для защиты нефтяного оборудования, модификаторами моторных топлив, эмульгаторами, исходными веществами для синтеза мономеров.
На основании полученных в ходе работы экспериментальных данных можно сделать вывод, что предложенный метод получения вышеупомянутого ненасыщенного ацетиленового диола, основанный на описанном Б.В. Иоффе способе [7] и реакции А.Е. Фаворского, осуществим в лабораторных условиях. Анализ полученных образцов продукта реакции, выполненный методом ЯМР-спектроскопии, подтверждает структуру полученного соединения. Предлагаемая в данной работе методика синтеза диметилдиэтилбутиндиола предпочтительней методики синтеза, описанной А.В. Щекуновым [6], поскольку более перспективна в технологическом плане.
Библиографическая ссылка
Меркулов В.В., Алмазов А.И., Мантлер С.Н. ПЕРСПЕКТИВНЫЙ МЕТОД СИНТЕЗА ДИМЕТИЛДИЭТИЛБУТИНДИОЛА // Успехи современного естествознания. 2017. № 10. С. 18-22;URL: https://natural-sciences.ru/en/article/view?id=36554 (дата обращения: 12.07.2026).