


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
QUANTITATIVE DETERMINATION OF LIDOCAINE AND BUPYUVACINE IN THE LIVER TISSUE BY THE METHOD OF CAPILLARY ELECTROPHORESIS

В последние годы в медицинской практике использование местных анестетиков стало довольно распространенным явлением. В первую очередь это связано с появлением местноанестезирующих лекарственных средств амидного типа (лидокаин, мепивакаин, бупивакаин, ропивакаин и др.), которые открыли новые возможности для повышения эффективности и безопасности контроля над болью. Кроме этого, развитие некоторых видов местной анестезии, таких как спинномозговая, эпидуральная и др., привело к увеличению числа хирургических операций на амбулаторных больных, а также к появлению новых, вызывающих меньшее кровотечение хирургических техник [1–3].
В то же время местные анестетики по частоте развития лекарственных осложнений стабильно занимают одно из первых мест. Побочные эффекты токсического характера развиваются, как правило, на фоне повышенных концентраций препарата в крови при использовании их в дозах, превышающих рекомендуемые, при случайном попадании в сосудистое русло или быстром введении препарата, а также индивидуальной непереносимости [1, 3].
При отравлении местноанестезирующими средствами паталогоанатомическая картина не характерна. Для выявления причины смерти лиц, принимавших анестетики, решающее значение приобретают результаты химико-токсикологического (судебно-химического) исследования. В то же время в клинической хирургической практике в ряде случаев возникает потребность проведения лабораторного анализа препарата в биологических средах [4]. В связи с этим актуальной задачей является разработка экспрессных, высокочувствительных и селективных методик анализа исследуемых соединений в биологических объектах.
Несмотря на значительное число работ, посвященных анализу местных анестетиков, продолжают оставаться актуальными вопросы исследования в области разработки новых и совершенствования существующих методик их химико-токсикологического анализа. Одним из современных и перспективных аналитических методов, отвечающих требованиям судебно-химической (химико-токсикологической) практики и клинической лабораторной диагностики, является капиллярный электрофорез (КЭ), характеризующийся сочетанием разделения веществ с последующей их детекцией в УФ-области спектра непосредственно в кварцевом капилляре. Метод отличается простым аппаратурным оформлением и доступными расходными материалами [4–6].
В связи с вышеизложенным целью исследования явилось изучение возможности количественного определения лидокаина и бупивакаина в ткани печени методом капиллярного электрофореза.
Исследования проводили с использованием системы капиллярного электрофореза «Капель-105» (ООО «Люмекс-Центрум», г. Санкт-Петербург). Ранее нами были установлены электрофоретические условия анализа исследуемых местных анестетиков (МА): рабочий электролит (РЭ) – буферный раствор Бриттона – Робинсона, рН 2,3; растворитель пробы (РП) – РЭ, разбавленный в 10 раз водой очищенной; ввод пробы – гидродинамическим способом (30 мбар х 15 сек); напряжение – +20 кВ; детектирование – при длине волны 200 нм; запись и обработка электрофореграмм (ЭФГ) – с помощью программного обеспечения «МультиХром для Windows» [4, 6, 7].
При разработке методики количественного определения МА методом КЭ готовили серию стандартных растворов МА в РП, содержащих от 0,5 до 20 мкг исследуемого вещества в 1 мл пробы и проводили электрофорез на приборе «Капель-105». Фотометрические сигналы (площади пиков) использовали для построения калибровочных графиков. При этом наблюдалась линейная зависимость величины фотометрического сигнала от концентрации МА в пробе. На основе полученных данных рассчитывали уравнения калибровочных графиков – y = 1,454x + 0,39 (r2 = 0,9991) для лидокаина и у = 1,085 х – 0,055 (r2 = 0,9997) для бупивакаина. Коэффициенты корреляции близки к единице, что подтверждает линейность методик в указанном диапазоне концентраций.
Для валидационной оценки методики количественного определения МА методом КЭ по уравнению калибровочного графика проводили электрофорез анестетиков с концентрацией 20, 10 и 1,0 мкг/мл в приведенных выше условиях. На ЭФГ определяли площади пиков и по уравнению калибровочного графика рассчитывали концентрации МА. При статистической обработке данных, полученных при количественном определении МА на трех уровнях концентраций (табл. 1, 2), отражается вполне удовлетворительная повторяемость и воспроизводимость результатов в пределах рекомендуемой аналитической области. Коэффициент вариации не превышает 1 %, а относительная ошибка среднего результата – 1,08 %.
Таблица 1
Воспроизводимость результатов методики количественного определения бупивакаина методом КЭ
|
Концентрация бупивакаина, мкг/мл |
Метрологические характеристики (n = 5) |
|||||
|
|
S |
СV % |
S |
Δ |
|
|
|
1,0 |
98,20 |
0,84 |
0,86 |
0,38 |
1,06 |
1,08 |
|
10,0 |
98,80 |
0,67 |
0,68 |
0,30 |
0,83 |
0,84 |
|
20,0 |
99,00 |
0,56 |
0,57 |
0,25 |
0,70 |
0,71 |



 %
%
Таблица 2
Воспроизводимость результатов методики количественного определения лидокаина методом КЭ
|
Концентрация лидокаина, мкг/мл |
Метрологические характеристики (n = 5) |
|||||
|
|
S |
СV % |
S |
Δ |
|
|
|
1,0 |
99,84 |
0,74 |
0,74 |
0,33 |
0,92 |
0,92 |
|
10,0 |
100,04 |
0,54 |
0,54 |
0,24 |
0,67 |
0,67 |
|
20,0 |
100,14 |
0,64 |
0,64 |
0,29 |
0,80 |
0,80 |



 %
%
Таблица 3
Сравнительная характеристика методов выделения МА из ткани печени
|
Экстрагент |
Внесено МА в 1,0 г печени, мкг |
Определено МА ( |
|
|
Лидокаин |
Бупивакаин |
||
|
% |
% |
||
|
Раствор щавелевой кислоты, рН 2,0 |
100,0 |
69,20 |
64,82 |
|
Раствор уксусной кислоты, рН 2,0 |
100,0 |
58,48 |
51,59 |
|
Раствор трихлоруксусной кислоты, рН 1,0 |
100,0 |
44,17 |
40,99 |
|
Контрольный опыт |
– |
– |
– |
 , %; n = 5)
, %; n = 5)Выделение и определение лекарственных средств в биологических объектах является одной из самых сложных задач судебно-химического (химико-токсикологического) и клинического лабораторного анализов. Трудность обусловлена изолированием микрограммовых количеств исследуемых соединений из биосубстратов и их очисткой от соэкстрактивных веществ [4, 7].
Предварительно нами были изучены условия экстракции анестетиков из водных растворов в зависимости от рН среды, природы органического растворителя, объема экстрагента и кратности экстрагирования. По результатам исследования установлено, что наибольший процент экстракции наблюдается с использованием в качестве экстрагента хлороформа при рН 10,0 (бупивакаин) и рН 9,0 (лидокаин), соотношении водной и органической фаз 1:1, кратности экстрагирования равной трем и составляет 94,71 % и 80,43 % для бупивакаина и лидокаина соответственно.
Также было проведено сравнительное изучение методов выделения искомых соединений с использованием извлекателей: раствора щавелевой кислоты, рН 2,0, раствора уксусной кислоты, рН 2,0 и раствора трихлоруксусной кислоты (ТХУК), рН 1,0. Исследование проводили по следующим методике: к 1 г мелкоизмельченной ткани печени добавляли 1 мл 0,01 % стандартных растворов МА, содержащих 100 мкг вещества в пробе, и перемешивали. Через 1 час к смеси добавляли 7 мл одного из вышеперечисленных извлекателей, перемешивали и оставляли на 1 час при периодическом перемешивании. Вытяжку центрифугировали (6000 об/мин, 10 мин). Надосадочную жидкость отделяли, добавляли 25 % раствор аммония гидроксида до рН 10,0 (бупивакаин) или рН 9,0 (лидокаин) по универсальной индикаторной бумаге и проводили жидкостное экстрагирование хлороформом (10 мл х 3, 10 мин). Хлороформные экстракты отделяли и оставляли в сухом защищенном от света месте для испарения. Сухие остатки при исследовании бупивакаина растворяли в 1,0 мл РП, при исследовании лидокаина – в 1 мл хлороформа и проводили реэкстрагирование 1 мл РП. Полученные реэкстракты центрифугировали (10000 об/мин, 10 мин) и количественно определяли методом КЭ в приведенных выше условиях.
Результаты исследований (табл. 3) показали, что наибольшая степень извлечения из ткани трупной печени наблюдается при использовании в качестве экстрагента раствора щавелевой кислоты, рН 2,0 и составляет 64,82 % для бупивакаина и 69,20 % для лидокаина.
При этом выбранные условия пробоподготовки позволяют эффектно провести очистку исследуемых оснований от соэкстрактивных веществ ткани печени (рисунок).
Для определения чувствительности методик количественного определения МА в биосубстрате к 1 г мелкоизмельченной ткани печени добавляли аликвоты стандартных растворов МА, содержащих определенное количество вещества в пробе. Дальнейшее выделение и определение осуществляли согласно разработанной методике. Результаты исследований представлены в табл. 4.
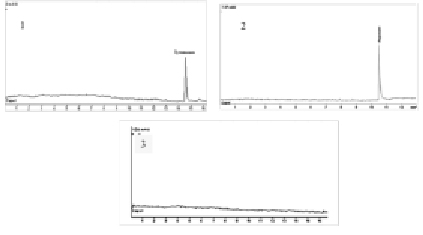
ЭФГ бупивакаина (1) и лидокаина (2), выделенных из ткани печени; контрольный опыт (3). Экстрагент – раствор щавелевой кислоты, рН 2,0 («Капель – 105»)
Таблица 4
Количественное определение МА в тканях трупной печени
|
№ п/п |
Внесено МА в 1 г ткани печени, мкг |
Определено МА |
|||||
|
Бупивакаин |
Лидокаин |
||||||
|
мкг |
% |
Метрологические характеристики |
мкг |
% |
Метрологические характеристики |
||
|
1 |
20,0 |
13,21 |
66,04 |
S = 0,97 СV = 1,47 % S
66,04 ± 1,20
|
13,84 |
69,20 |
S = 1,08 СV = 1,55 % Sx = 0,48
69,20 ± 1,34
|
|
2 |
1,0 |
0,656 |
65,60 |
S = 0,55 СV = 0,84 % S
65,60 ± 0,70
|
0,67 |
67,29 |
x = 67,29 S = 0,31 СV = 0,47 % S
67,29 ± 0,40
|
|
3 |
0,5 |
0,316 |
63,20 |
S = 1,10 CV = 1,74 % S
63,20 ± 1,36
|
0,33 |
66,52 |
x = 66,52 S = 0,45 СV = 0,68 % S
66,52 ± 0,56
|
|
4 |
0,1 |
+ |
– |
– |
+ |
– |
– |
 = 66,04
= 66,04 = 0,43
= 0,43 ± Δ
± Δ =
= = 1,82 %
= 1,82 % = 69,20
= 69,20 ± Dx =
± Dx = = 1,93 %
= 1,93 % = 65,60
= 65,60 = 0,25
= 0,25 ± Δ
± Δ =
= = 1,07 %
= 1,07 % = 0,14
= 0,14 ± Dx =
± Dx = = 0,59 %
= 0,59 % = 63,20
= 63,20 = 0,49
= 0,49 ± Δ
± Δ =
= = 2,15 %
= 2,15 % = 0,20
= 0,20 ± Dx =
± Dx = = 0,84 %
= 0,84 %Примечание. (+) – количество вещества ниже предела количественного определения.
Таким образом, разработанные методики количественного определения лидокаина и бупивакаина в ткани печени характеризуются удовлетворительной повторяемостью в пределах данной аналитической области по критерию воспроизводимости (стандартное отклонение среднего результата) и позволяют определить 63,20–66,04 % бупивакаина и 69,20–66,52 % лидокаина. При этом граница определения составила 0,5 мкг, а граница обнаружения – 0,1 мкг обоих веществ в 1 г ткани печени.
Выводы
1. Проведена сравнительная характеристика методов выделения исследуемых местных анестетиков из ткани печени. При этом наибольшая степень выделения наблюдается при использовании в качестве извлекателя раствора щавелевой кислоты, рН 2,0. Граница определения составила 0,5 мкг, а граница обнаружения – 0,1 мкг исследуемых веществ в 1,0 г ткани печени.
2. Разработанные методики количественного определения лидокаина и бупивакаина в ткани печени являются валидными по критерию повторяемости результатов измерений в пределах рекомендуемых аналитических областей.
3. Предложенные методики выделения и определения исследуемых местных анестетиков высокочувствительны, селективны и могут быть рекомендованы для проведения судебно-химического (химико-токсикологического) и клинического лабораторного анализов.
Библиографическая ссылка
Смирнова А.В., Фомин А.Н., Семёнов М.Б., Каджоян Л.В. КОЛИЧЕСТВЕННОЕ ОПРЕДЕЛЕНИЕ ЛИДОКАИНА И БУПИВАКАИНА В ТКАНИ ПЕЧЕНИ МЕТОДОМ КАПИЛЛЯРНОГО ЭЛЕКТРОФОРЕЗА // Успехи современного естествознания. 2017. № 12. С. 16-20;URL: https://natural-sciences.ru/en/article/view?id=36599 (дата обращения: 02.07.2026).