


Scientific journal
Advances in current natural sciences
ISSN 1681-7494
"Перечень" ВАК
ИФ РИНЦ = 0,976
ELECTRONIC STRUCTURE OF NEW CRYSTALS LIMS2 (M = B, AL, GA, IN)

Поиск новых материалов для нелинейной оптики среднего ИК-диапазона и развитие технологий синтеза все более сложных по составу и структуре веществ стимулировали получение и экспериментальное исследование Li-содержащих соединений, принадлежащих семейству хорошо изученных к настоящему времени алмазоподобных соединений I–III–VI2. Повышенный интерес к кристаллам LiMX2 (M = B, Al, In, Ga; X = O, S, Se, Te) вызван наличием ряда физических и физико-химических свойств (относительно большая ширина запрещенной зоны, высокий коэффициент теплопроводности, низкая анизотропия линейного теплового расширения и др.), делающих кристаллы LiMX2 более перспективными по сравнению с их кристаллохимическими аналогами, например, CuMX2 и AgMX2. Благодаря проведенным за последние десять лет исследованиям был накоплен большой массив экспериментальных данных, полученных в нашей стране группой Л.И. Исаенко из Новосибирска [1–3] и их зарубежными коллегами, осуществившими успешное выращивание и изучение свойств тройных Li-содержащих халькогенидов [4–6]. Теоретические исследования из первых принципов электронной структуры и колебательных свойств кристаллов LiMX2 появились сравнительно недавно и посвящены изучению кристаллов в структурах β-феррита натрия (орторомбическая фаза типа β-NaFeO2) [7, 8] или халькопирита (тетрагональная фаза типа CuFeS2) [9–11]. Электронное и колебательное строение кристаллов LiMS2, которые обычно кристаллизуются в орторомбической фазе, частично изучены и полученные из эксперимента или с помощью теоретических расчетов данные представлены в перечисленных выше работах. Что касается тетрагональной фазы, то какие-либо сведения о синтезе или компьютерном моделировании кристаллов LiMS2 со структурой халькопирита в литературе отсутствуют.
Цель исследования: моделирование и исследование с использованием методов теории функционала плотности энергетической зонной структуры и химической связи в алмазоподобных кристаллах LiMS2 (M = B, Al, Ga, In) с решеткой халькопирита.
Материалы и методы исследования
Подобие орторомбической и тетрагональной кристаллических структур, характерных для кристаллов LiMX2, было подробно рассмотрено и проанализировано в работе [12] на примере реальных кристаллов LiInSe2, синтезированных в структурах халькопирита и β-феррита натрия. В настоящей работе мы моделируем кристаллы LiMS2 (M = B, Al, Ga, In) в структуре халькопирита (рис. 1), которая является производной от структуры сфалерита (увеличенной вдвое по оси z из-за наличия катионов двух сортов) и характеризуется параметрами решетки a и c, их отношением γ = c/a (тетрагональное сжатие) и смещением анионов из узлов гранецентрированной подрешетки, определяемым их координатой x. Элементарная ячейка халькопирита содержит 8 атомов с координатами: Li – (0,0,0) (0,½,¼); M – (0,0,½) (0,½,–¼); S – (x,¼,1/8) (–x,¼,1/8) (–¼,x,–1/8) (¼,-x,-1/8), в единицах a(1,1,γ).
В структуре халькопирита катионы (Li, M) окружены четырьмя эквивалентными анионами S, а каждый анион S окружен парой атомов Li и парой атомов сорта M, таким образом, что формируются катионные LiS4, MS4 и анионные Li2SM2 тетраэдры.
Постоянные решетки a, c и смещение анионов x из узлов ГЦК подрешетки, определяющие размер кристаллической ячейки и положение анионов в структуре халькопирита, были вычислены по формулам из работы [13]. Полученные таким образом значения a, c и x путем оптимизации геометрии кристалла были приведены к равновесным значениям, которые представлены в таблице и использовались нами в расчетах электронного строения кристаллов LiMS2 (M = B, Al, Ga, In).
Параметры кристаллической структуры кристаллов LiMS2
|
Параметры |
LiBS2 |
LiAlS2 |
LiGaS2 |
LiInS2 |
|
a, Å |
5,4632 |
5,6770 |
5,6902 |
5,8090 |
|
c, Å |
8,1464 |
10,1316 |
10,3749 |
11,2801 |
|
x |
0,3249 |
0,2797 |
0,2744 |
0,2501 |
|
γ |
1,491 |
1,785 |
1,823 |
1,942 |
|
RLi-S, A |
2,460 |
2,478 |
2,479 |
2,492 |
|
RM-S, A |
1,954 |
2,276 |
2,314 |
2,491 |
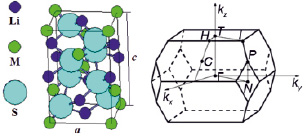
Рис. 1. Кристаллическая ячейка и зона Бриллюэна соединений LiMS2 (M = B, Al, Ga, In) со структурой халькопирита
Кристаллическая ячейка кристаллов LiMS2 (M = B, Al, Ga, In) со структурой халькопирита (пространственная группа № 122, 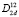 или I-42d) содержит две формульные единицы с четырьмя неэквивалентными атомами и является сильно деформированной, так как тетрагональное сжатие γ = c/a кристаллов LiMS2 (M = B, Al, Ga) много меньше «идеального» значения γ = 2,00. Параметр смещения анионов (S) для кристаллов LiMS2 (M = B, Al, Ga) также сильно отличается от «идеального» x = 0,25, что обусловлено существенным различием длин связей Li-S и M-S: от 21 % в LiBS2 до 7 % в LiGaS2. Исключение представляет кристалл LiInS2, в котором длины связей практически равны, и соответственно, кристаллические параметры a, c и x близки к идеальным.
или I-42d) содержит две формульные единицы с четырьмя неэквивалентными атомами и является сильно деформированной, так как тетрагональное сжатие γ = c/a кристаллов LiMS2 (M = B, Al, Ga) много меньше «идеального» значения γ = 2,00. Параметр смещения анионов (S) для кристаллов LiMS2 (M = B, Al, Ga) также сильно отличается от «идеального» x = 0,25, что обусловлено существенным различием длин связей Li-S и M-S: от 21 % в LiBS2 до 7 % в LiGaS2. Исключение представляет кристалл LiInS2, в котором длины связей практически равны, и соответственно, кристаллические параметры a, c и x близки к идеальным.
Теория функционала плотности – DFT (density functional theory) хорошо себя зарекомендовала в исследованиях электронного строения различных по составу кристаллических твердых тел и составляет основу большинства современных программных кодов. Мы применили данную теорию с целью реализации модельных расчетов из первых принципов (ab initio) для группы гомологических кристаллов LiMS2 (M = B, Al, Ga, In). Оптимизация геометрии и вычисления зонной структуры E(k), плотности состояний N(E) и распределения электронной плотности ρ(r) проводились в рамках теории функционала плотности с использованием программного кода CRYSTAL [14] и заложенного в нем гибридного метода ab initio расчета B3LYP, который, в частности, считается одним из лучших при вычислении ширины запрещенной зоны. Высокая точность расчетов и сходимость по полной энергии обеспечивались разбиением зоны Бриллюэна (рис. 1) на сетку из специальных точек 16×16×16 и обрывом энергии при 40 ридбергах.
Результаты исследования и их обсуждение
Зонные спектры E(k) исследуемых кристаллов LiMS2 (M = B, Al, Ga, In), вычисленные в наиболее характерных для зоны Бриллюэна халькопирита (рис. 1) точках высокой симметрии T = (001), Г = (000), N = (½½0), а также вдоль соединяющих их линий в единицах 2π/a(1,1,1/γ), представлены на рис. 2.
За начало шкалы энергий выбрана вершина валентной зоны кристалла LiBS2, серым цветом выделена запрещенная зона Eg, значения которой приведены на рис. 1. Цифрами (1, 4, 5) на рисунке вблизи краев зоны проводимости (c) и валентной зоны (v) обозначены соответствующие неприводимые представления (Γ1C, Γ4V, Γ5V).
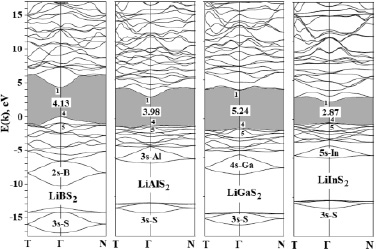
Рис. 2. Зонная структура кристаллов LiMS2 (M = B, Al, Ga, In)
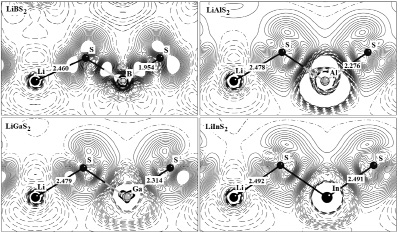
Рис. 3. Деформационная плотность Δρ(r) кристаллов LiMS2 (M = B, Al, Ga, In)
В целом валентная зона кристаллов LiMS2 (M = B, Al, Ga, In) имеет типичный для кристаллов LiMX2 со структурой халькопирита вид [11, 12]. Можно видеть три разрешенные подзоны, разделенные двумя запрещенными участками энергии, как в кристаллах LiGaS2 и LiInS2. В кристаллах LiBS2 и LiAlS2 эти подзоны можно только проследить, так как две верхние подзоны перекрываются между собой. Причиной такого перекрывания является сильное тетрагональное сжатие (γ < 1,8), как в случае LiBO2 [11]. Во всех кристаллах LiMS2 нижняя подзона из четырех уровней энергии содержит преимущественно вклады 3s-состояний атомов S. Следующая подзона из двух энергетических уровней включает преобладающие вклады s-состояний атомов M (Ga, In). В кристаллах LiBS2 и LiAlS2 эта подзона состоит из четырех уровней энергии, которые вплотную прилегают к верхней подзоне, состоящей из десяти энергетических уровней, образованных в основном из p-состояний атомов S. Полная ширина валентной зоны ΔEV уменьшается от ~16 до 12 эВ в ряду LiBS2>LiAlS2>LiGaS2>LiInS2. Рассмотрение изоанионного ряда сульфидов LiMS2 показало, что при замещении катионов B>Al>Ga>In (рис. 2) прослеживаются отделение второй связки зон от верхней связки валентных зон и нелинейное изменение структуры валентных зон, которые коррелируют с нелинейным изменением ковалентных радиусов (rM) и электроотрицательностей (χ), равных соответственно: B(0,89, 1,4)→Al(1,34, 1,35)→ Ga(1,29, 1,5)→In(1,49, 1,6).
Экстремумы валентной зоны и зоны проводимости кристаллов LiMS2 реализуются в центре зоны Бриллюэна (точка Г). Поскольку абсолютный минимум зоны проводимости (Γ1C – неприводимое представление) и абсолютный максимум валентной зоны (Γ4V) расположены в одной точке зоны Бриллюэна, а прямой переход Γ4V>Γ1C разрешен, то исследуемые кристаллы являются прямозонными. Разность между этими уровнями определяет ширину запрещенной зоны Eg, значения которой изменяются следующим образом (рис. 2): 4,13 (LiBS2), 3,98 (LiAlS2), 5,24 (LiGaS2), 2,87 (LiInS2) эВ. Кристаллическое расщепление, равное разности между двумя верхними уровнями вершины валентной зоны ΔКР = E(Γ4V)-E(Γ5V) убывает в изоанионном ряду с увеличением атомного номера катиона M (B, Al, Ga, In): 0,85 (LiBS2), 0,23 (LiAlS2), 0,21 (LiGaS2), 0,09 (LiInS2) эВ.
Из расчетов парциальных плотностей состояний N(E) установлено, что дно зоны проводимости содержит в основном p-состояния атомов S, с примесью s-состояний атомов M (B, Al, Ga, In), как в случае других орторомбических и тетрагональных кристаллов LiMX2, рассмотренных в работах [11, 12].
Вычислены полные ρ(r) и деформационные Δρ(r) плотности распределения заряда валентных электронов и исследованы особенности образования химической связи в кристаллах LiMS2 (M = B, Al, Ga, In) со структурой халькопирита. Деформационная плотность исследуемых кристаллов получается в результате вычитания из кристаллической электронной плотности ρ(r) суммы электронных плотностей атомов, входящих в состав кристалла. На рис. 3 представлены карты распределения деформационной Δρ(r) электронной плотности кристаллов LiMS2 (M = B, Al, Ga, In) со структурой халькопирита в плоскости содержащей атомы разного сорта. Анализ карт распределения полной электронной плотности показывает, что основной заряд сосредоточен на анионе (S). Такое распределение типично и для других кристаллов LiMX2 с различной структурной модификацией [11, 12].
На картах деформационной плотности Δρ(r) хорошо видны локализованные на связях Li-S и M-S заряды, характеризующие ковалентную составляющую химической связи между катионами и анионами. Связи Li-S являются в большей степени ионными и менее прочными, чем связи M-S, что можно проследить по картам на рис. 3. Увеличение длин связей свидетельствует об их ослаблении в ряду LiBS2>LiAlS2>LiGaS2>LiInS2.
Заключение
Изучение электронного строения изоэлектронного ряда LiBS2→LiAlS2→ LiGaS2→LiInS2 в рамках единого подхода на основе теории функционала плотности обеспечило возможность получить достоверные данные об энергетической зонной структуре и особенностях формирования химической связи, обусловленных изменением химического состава в исследуемых кристаллах. Получены равновесные структурные параметры четырех новых кристаллов со структурой халькопирита. Все кристаллы LiMS2 (M = B, Al, Ga, In) являются широкозонными Eg > 2,5 эВ, что делает их перспективными материалами для практического применения в оптоэлектронике и устройствах, работающих в среднем ИК-диапазоне, как большинство кристаллов LiMX2 с различной структурной модификацией. Полученные карты распределения деформационной электронной плотности в кристаллах LiMS2 (M = B, Al, Ga, In) хорошо иллюстрируют особенности ионно-ковалентной связи в них. Результаты расчетов зонной структуры и деформационной плотности Δρ(r) не противоречат данным, полученным ранее для других кристаллов семейства халькопирита. Первопринципное моделирование кристаллической структуры, установленные из расчетов особенности электронного строения, количественные характеристики энергетической зонной структуры, тип химической связи и выявленное влияние химического состава в рассмотренных кристаллах LiMS2 (M = B, Al, Ga, In) на их свойства, могут быть полезны с научной точки зрения. Они также имеют практическое значение для специалистов, занимающихся поиском новых полупроводниковых материалов с уникальными оптическими свойствами и подбирающих условия их синтеза в структуре халькопирита, которая, согласно расчетам, является вполне реальной и может быть получена при высоких температурах как фаза высокого давления.
Библиографическая ссылка
Басалаев Ю.М., Дугинов Е.В., Дугинова Е.Б. ЭЛЕКТРОННОЕ СТРОЕНИЕ НОВЫХ КРИСТАЛЛОВ LIMS2 (M = B, AL, GA, IN) // Успехи современного естествознания. 2018. № 11-1. С. 12-17;URL: https://natural-sciences.ru/en/article/view?id=36898 (дата обращения: 02.07.2026).