INTRAVENOUS IMMUNOGLOBULINS IN THE VIEW OF QUALITY, EFFICACY AND SAFETY PROFILES
1
Supotnitskiy M.V. 1, Elapov A.A. 1, Borisevich I.V. 1, Kudasheva E.Y. 1, Klimov V.I. 1, Lebedinskaya E.V. 1
1 Federal State Budgetary Institution «Scientific Center for Expertise of Medical Application Products» of the Ministry of Health of the Russia
Intravenous immunoglobulin (IVIG) preparations are polyclonal IgG antibodies, obtained from donor blood plasma. Modern IVIG have high IgG purity with normal subclass distribution and the monomer and dimer content of more than 95 % of the total volume of a preparation. The activity of Fc-fragment of IgG molecule reaches almost 100 %. The article describes the evolution of the approaches to obtaining IVIG, safety measures and quality control in the production of IVIG and the characteristics of various IVIG generations and quality parameters. Quality and safety issues of IVIG are resolved under strict state regulation of the processes of collection and fractionation of donor plasma and monitoring of manufacture. Resolving the issues specific for IVIG is carried out by a thorough purifying of an IVIG preparation form immunoglobulin aggregates, proteases, plasmin, plasminogen, prekallikrein activator, impurities of IgA and IgM. Viral safety is achieved by a multi-step manufacturing process that includes at least two independent methods (solvent-detergent treatment + incubation at low pH or pasteurization, combined with the polyethylene glycol processing). It is assumed that for today the development limit for the whole field of obtaining IVIG from donor plasma has been achieved. Certain improvements will be related to upgrading the efficiency of manufacturing technologies and methods of IVIG clinical applications (preparation for subcutaneous administration, combination of different immunoglobulin preparations, etc.), their viral safety, methods of eliminating the impurities from the components that have previously not been considered to be able to influence the outcome of clinical use (soluble molecules CD4, CD8, HLA, thrombin, trace amounts of blood clotting factors VIII, IX, X, XI, XII and others). However, up till now the technologies ensuring viral safety have not been developed a 100 % yet. Any combination of these factors can provide only «maximum level of viral safety» in the obtaining of IVIG. It is also not possible with the existing approach to obtaining IVIG (as a pool of IgG with a high purity derived from thousands of donors plasma) to achieve reproducible results and their clinical application. The unresolved problems limits clinical use of IVIG and their further improvement. At the same time another type of immunoglobulin preparations are assumed to appear on the market: genetically engineered preparations that are well-characterized in terms of molecular composition, having a high selectivity for the target exposure.
intravenous immunoglobulin
Fc-fragment
IgG
polyclonal antibody
monomeric IgG
measles
immunoglobulins for intramuscular administration
idioagglutinin to antigens A
B
D
HIV-1
HIV-2
hepatitis A
B and C
parvovirus B19
HBsAg
cryoprecipitate
sepsis
WHO
Иммуноглобулины для внутривенного введения (ВВИГ) являются наиболее часто используемыми препаратами, изготовленными из плазмы крови доноров. Государственный реестр лекарственных средств и Анатомо-терапевтическо-химическая (Anatomical Therapeutic Chemical) классификация (АТХ) относят ВВИГ к фармацевтической группе «медицинские иммунобиологические препараты» (код по АТХ JO6BA02) [3].
Современные ВВИГ получают фракционированием плазмы крови человека. Они представляют собой препараты поликлональных антител класса IgG, синтезированных В-лимфоцитами в ответ на антигенные стимулы, имевшие место на протяжении жизни человека-донора. IgG – гликопротеин с молекулярной массой около 150 кДа, содержащийся в плазме человека в количестве от 7 до 12 г⁄л [12]. Класс IgG классифицируют на четыре подкласса (IgG1, IgG2, IgG3, IgG4), класс IgA – на два подкласса (IgA1, IgA2). Все классы и подклассы составляют девять изотипов, которые присутствуют в норме у всех индивидов. Каждый изотип IgG определяется последовательностью аминокислот константной области тяжелой цепи [2].
Современные препараты ВВИГ подразделяются на три группы [3]:
I. Стандартные препараты – содержат в основном IgG (иммуноглобулин человека нормальный для внутривенного введения).
II. Стандартные специфические (гипериммунные) препараты – содержат в основном IgG, но имеют более высокое содержание противовирусных антител.
III. Обогащенные препараты ВВИГ – содержат антитела классов IgG, IgM, IgA против патогенных вирусов и бактерий.
Эффективность и безопасность медицинского применения ВВИГ определяются дуализмом функции IgG: они могут специ фически взаимодействовать с чужеродными антигенами и одновременно способны вызывать неспецифические эффекты. Такая функциональная дихотомия является следствием особенностей структуры молекулы IgG. Ее вариабельный регион (два Fab-фрагмента) состоит из легкой и частично из тяжелой цепей и специфически взаимодействует с антигенами, что обусловлено меняющейся от белка к белку последовательностью аминокислотных остатков в N-концевой части молекулы. Константный регион (Fc- или кристаллизующийся фрагмент) связывает компонент комплемента 1 (С1) и взаимодействует с Fc-рецепторами макрофагов или нейтрофилов. Активация эффекторной функции Fc-фрагмента антитела происходит после агрегации IgG на поверхности антигена, структура молекулы меняется, что служит сигналом для запуска системы комплемента или индукции опсонизации через фагоцитоз (рис. 1) [2].
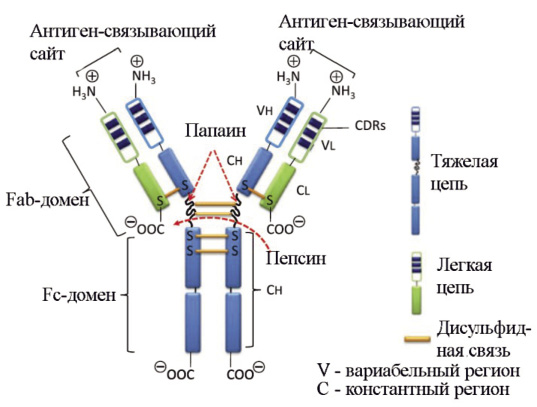
Рис. 1. Схематическое строение иммуноглобулина G. Обработка пепсином приводит к расщеплению в участке молекулы на С-концевой стороне, за дисульфидной связью, соединяющей две тяжелые цепи вариабельного участка IgG. В результате образуются один сдвоенный F(аb’)-фрагмент [F(аb’)2] и один Fc-фрагмент. Расщепление IgG папаином происходит в N-концевом участке, непосредственно перед дисульфидной связью, в результате образуются два одинаковых Fab-фрагмента и один Fc-фрагмент [12]
Обобщение опыта клинического применения ВВИГ позволило Е.К. Донюш [3] утверждать, что они обеспечивают:
– увеличение бактерицидной активности сыворотки, стимуляцию фагоцитоза, нейтрализацию некоторых бактериальных токсинов;
– блокаду дифференцировки В-лимфоцитов, продуцирующих антитела;
– предотвращение или блокаду взаимодействия аллергена с IgE, фиксированного на тучной клетке, за счет IgG4 блокирующих антител;
– подавление продукции аллерген-специфических и ауто-антител за счет воздействия антиидиотипических антител;
– снижение продукции и активности провоспалительных цитокинов;
– предупреждение комплемент-зависимого повреждения тканей за счет связывания C3b и C4b компонентов комплемента;
– предохранение от дополнительных вирусных инфекций, обладающих триггерным эффектом при аутоиммунных заболеваниях.
Первыми неспецифическими иммуноглобулинами, использованными в клинической практике, были иммуноглобулины для внутримышечного применения (intramuscular immunoglobulin, IMIG). В России разрешены иммуноглобулин человека нормальный, противоаллергический и 6 специфических иммуноглобулинов, получаемых из плазмы крови иммунизированных людей (противооспенный, антирабический, антистафилококковый, противостолбнячный, против гепатита В и клещевого энцефалита) [1].
Технология приготовления таких иммуноглобулинов разработана в 1940-х гг. Включает этапы получения плазмы крови человека и осаждения из нее IgG этанолом при температуре ниже 0 °С и определенном значении рН [1, 2]. Дополнительной очистки IgG не проводится. Получаемый препарат содержит 70–80 % мономерных IgG и значительные количества IgA и IgM. Вводимые в его составе в организм человека антитела имеют обычный период полураспада, активируют комплемент в присутствии антигена и обладают опсонизирующими свойствами. Применение нормальных иммуноглобулинов оказалось эффективным для профилактики и лечения кори, гепатита А и для предупреждения бактериальных инфекций у детей с наследственной агаммаглобулинемией [3, 12].
Непреодолимыми в рамках данной технологии получения IgG недостатками данных препаратов, стали болезненность в месте введения, низкая скорость поступления антител в системный кровоток и невозможность быстро создавать высокие концентрации антител в ургентных ситуациях. При попытках внутривенного введения у пациентов развивались опасные анафилактоидные реакции и гипотония, что связано с неспецифической активацией комплемента в результате спонтанного образования агрегатов иммуноглобулинов и наличием в препарате следовых количеств протеаз [7]. Поэтому применение препаратов, полученных по данной технологии, ограничено внутримышечным введением.
Попытки повысить безопасность ВВИГ в 1960-х гг. привели к значительному снижению терапевтической эффективности таких препаратов. Сделав правильный вывод о ключевой роли Fc-фрагмента IgG в неспецифической активации комплемента, разработчики предприняли попытки получить ВВИГ либо без такого фрагмента, либо инактивировав его в составе IgG. Для этого IgG расщепляли пепсином, но F(аb’)2 исчезали из кровотока в течение 2 суток; расщепление плазмином приводило к получению моновалентных F(аb’) с низкой нейтрализующей способностью; не удавалось добиться и полной инактивации Fc-фрагмента в составе IgG обработкой алкилирующими и ацетилирующими соединениями. К тому же энзиматические и химические модификации IgG приводили к утрате ими важной физиологической функции – активации комплемента комплексом «антиген-антитело», что необходимо для эффективного лизиса бактерий и вирусов, поглощенных лейкоцитами [12]. В настоящее время выпуск таких препаратов прекращен [2].
Низкая терапевтическая эффективность энзиматических и химических производных IgG вынудила разработчиков ВВИГ в начале 1970-х гг. вернуться к получению интактного IgG. Особую значимость для потребителей препаратов крови в те годы приобрела их вирусная безопасность. Проблемы качества и безопасности ВВИГ на основе интактного IgG решались строгой государственной регламентацией процессов сбора и фракционирования плазмы доноров, контроля производства. Были разработаны национальные и международные документы, регулирующие производство ВВИГ. Система таких мер приведена в табл. 1.
Таблица 1
Меры безопасности и контроля качества при производстве ВВИГ [12]
|
Производственный этап
|
Требования к этапу, критические для обеспечения качества/безопасности ВВИГ
|
|
Учреждение по заготовке крови (лицензирование и проверяются национальным регулирующим органом; контроль оборудования, фракционирующего плазму)
|
|
Скрининг донор крови и плазмы
|
Эпидемиологический надзор за населением, идентификация доноров, конфиденциальное анкетирование кандидатов в доноры на наличие факторов риска, анализ их медицинских документов и анкеты
|
|
Процедура сбора крови/плазмы
|
Контроль длительности процедуры забора крови у доноров, смешивания с растворами, предотвращающими коагуляцию, температуры от момента забора крови до ее направления в блок переработки и др. параметров процесса, определенных нормативным документом
|
|
Тестирование донора на вирусоносительство перед забором крови
|
Выявление антител к ВИЧ 1 и 2, вирусам гепатитов А, В и С, HBsAg, парвовируса В19. Исследование должно проводится индивидуально или минипулами, использованные методы должны быть валидированы
|
|
Другие тесты у доноров
|
Тестирование на изоагглютинины к антигенам А, В, D, не использование крови доноров с высокими титрами антител к этим антигенам
|
|
Переработка крови/плазмы
|
Должна использоваться плазма, замороженная в течение 24–72 ч после забора
|
|
Замораживание и хранение плазмы
|
Должен использоваться быстрый способ замораживания плазмы, в процессе ее хранения температура не должна меняться
|
|
Транспортировка плазмы
|
Во время транспортировки должен вестись постоянный мониторинг температуры ее хранения с записью соответствующим оборудованием. Температура хранения при транспортировке должна быть минус 20 °С или менее
|
|
Предприятия по фракционированию плазмы крови (лицензирование и инспекция национальным регулирующим органом)
|
|
Тестирование мини-пулов
|
Используются технологии амплификации нуклеиновых кислот. Определяется нуклеиновая кислота ВИЧ 1 и 2, вирусов гепатитов А, В и С, парвовируса В19
|
|
Тестирование производственного пула
|
Антитела на ВИЧ 1 и 2, вирус гепатита С, HBsAg (обязательно); РНК вируса гепатита С (обязательно в Европе). Исследование нуклеиновых кислот других вирусов – в соответствии с регулирующими документами
|
|
Предприятие по фракционированию плазмы
|
Должно быть разработано, построено и функционировать в соответствии с GMP
|
|
Этапы очистки белков и инактивация вирусов
|
Все процессы должны быть валидированы, все операции должны выполняться в соответствии с утвержденной СОП
|
|
Стерилизующая фильтрация и асептическое заполнение упаковок
|
То же
|
|
Лиофильное высушивание (когда необходимо)
|
То же
|
|
Проверка конечного продукта
|
Все операции должны выполняться в соответствии с утвержденной СОП
|
Устранение недостатков, характерных для ВВИГ на основе интактного IgG, проводилось путем более тщательной очистки препарата от агрегатов иммуноглобулинов, протеаз, плазмина, плазминогена, активатора прекалликреина, примесей IgA и IgM. Е.К. Донюш [3] выделяет 4 поколения ВВИГ.
Препараты первого поколения – начало 1970-х гг., это энзиматически и химически модифицированные IgG, не имевшие функционального Fc-фрагмента.


Рис. 2. Основные технологии, используемые для получения
коммерческого ВВИГ и других компонентов крови из плазмы человека [12].
А, Б, В и Г – технологии, описанные в работах Bertolini J. [8], Teschner
W. et al. [17], Terpstra F.G. et al. [16] и Stucki M. еt al. [15]
соответственно
Препараты второго поколения – конец 1970-х гг., включали полностью интактную молекулу IgG с активной Fc-функцией и могли применяться не только с целью заместительной, но и иммуномодулирующей терапии. Однако степень их очистки оставалась низкой, они содержали IgA в количествах, вызывающих анафилактические реакции при внутривенном введении, показатель Fc-функции не превышал 70–75 %.
Препараты третьего поколения создавались в середине-конце 1980-х гг., характеризовались высокой чистотой и полной активностью Fc-фрагмента, высокой степенью вирусной безопасности, достигаемой многоступенчатым процессом производства. Выпускались в жидком виде и могли храниться при температуре 2–8 °С.
Препараты четвертого поколения – препараты, удовлетворяющие более жестким требованиям вирусной безопасности и физиологического распределения IgG по подклассам. Разработаны в 1990-х гг. и широко используются в настоящее время. Имеют высокую чистоту IgG с нормальным распределением по подклассам, содержание мономеров и димеров более 95 %. Активность Fc-фрагмента молекулы IgG приближается к 100 %. Препараты получают, используя многоступенчатую схему инактивации вирусов, включающую не менее двух самостоятельных методов (сольвент-детергентная обработка + инкубация при низких значениях рН или пастеризация в сочетании с обработкой полиэтиленгликолем). Препараты выпускают в жидком виде, допускается хранение при комнатной температуре.
В качестве стабилизаторов ВВИГ четвертого поколения используются вещества, безопасные для пациентов с нарушением углеводного обмена и дисфункцией почек, 10 %-е растворы ВВИГ позволяют снизить объемную нагрузку на пациента. Учитывая, что степень очистки IgG в препаратах четвертого поколения приближается к 100 %, их можно считать пределом развития всего направления получения ВВИГ из плазмы крови доноров. Отдельные усовершенствования будут касаться повышения эффективности технологий получения и клинического применения ВВИГ (препарат для подкожного применения, комбинации различных иммуноглобулинов в препарате и др.), их вирусной безопасности, способов очистки от примесей компонентов, которые раньше не считали способными влиять на результат клинического применения (растворимые молекулы CD4, CD8, HLA, следовые количества факторов свертывания крови VIII, IX, X, XI, XII и др.). Основные технологии, используемые в настоящее время для получения ВВИГ и других компонентов крови, показаны на рис. 2.
Получение коммерческого ВВИГ из плазмы человека начинается с отделения от нее так называемого «криопреципитата». Для этого замороженную плазму оттаивают в контролируемых условиях, затем центрифугируют с помощью центрифуг непрерывного действия (при температуре 2–3 °С). Полученный криосупернатант («cryosupernatant» или «cryo-poor plasma») используют для хроматографического выделения белков протромбинового комплекса, антитромбина или C1-ингибитора системы комплемента крови. Для осаждения фибриногена супернатант может подвергаться первой этанольной преципитации (8 % этанола при нейтральном значении pH). Для выделения ВВИГ используют три или четыре этапа осаждения этанолом при низких значения рН (осаждение по Кону) и обработку пепсином в низких концентрациях – при этом получают так называемую фракцию II (fraction II). Для уменьшения антикомплементарной активности ВВИГ и более глубокой очистки от IgА, осаждение этанолом может быть дополнено комбинацией этапов хроматографической очистки на катионных и анионных обменниках (с заменой третьего и четвертого осаждения по Кону). Выход фракции II при таком способе очистки плазмы составляет 3–6 г/л, в зависимости от исходного количества IgG в плазме и использованных методов его преципитации и последующей очистки [12].
Стандартная технология производства позволяет из 1 литра плазмы получить до 2,5 упаковок альбумина 10 %, до 3,5 упаковок иммуноглобулина для внутривенного введения 5 % и около 200–250 МЕ фактора VIII [4].
Основной тенденцией в стабилизации препаратов ВВИГ в настоящее время считается использование высокой концентрации IgG (100 мг/ мл по белку); слабокислой среды (рН 4,5–5,5); включение в лекарственную форму стабилизаторов, таких как полиолы (сорбит), сахара (мальтоза, глюкоза), или аминокислоты (глицин, пролин, изолейцин); отсутствие в препаратах хлорида натрия и сахарозы; осмолярность, близкая к физиологической; отсутствие консервантов и антибиотиков [1, 10].
Требования к свойствам ВВИГ следующие [3]:
– они должны иметь оптимальный спектр антител в соответствии с инфицированностью населения (более 1000 доноров);
– обладать доказанной эффективностью (с помощью контролируемых клинических исследований);
– распределения IgG по подклассам должно соответствовать их содержанию в плазме крови;
– для каждой партии препарата должен быть задекларирован титр антител;
– макроагрегаты должны составлять менее 1 % общего содержания IgG;
– антикомплементарная активность не должна превышать 1,0 СН50/1 мг белка протеина;
– гемолизины не должны содержаться в препарате, титр АВ-антител должен быть менее 1:8;
– активаторы прекалликреина, консерванты, активированные ферменты, токсические вещества не должны присутствовать в препарате;
– если предусмотрено применение у пациентов с врожденным дефицитом IgA; содержание IgA должно быть минимальным;
– высокая противовирусная очистка.
Возможность получения IgG с высокой степенью очистки позволила в последнее десятилетие вернуться к практике их подкожного введения, оказавшейся неудачной в 1940–1950-е гг. из-за большого количества реакций на балластные компоненты иммуноглобулинов, получаемых по технологиям того времени. Иммуноглобулины для подкожного введения (subcutaneous immunoglobulin, SCIG) в основном используются для лечения пациентов с врожденными нарушениями антителообразования (низкий уровень IgA), с повышенным содержанием в сыворотке крови воспалительных маркеров, флебитами, заболеваниями почек и другой патологией, создающей условия для осложнения при введении ВВИГ [6]. В настоящее время за рубежом в клинической практике используется не менее 6 SCIG. Сравнение свойств SCIG с аналогичными свойствами ВВИГ приведено в табл. 2.
Для улучшения проницаемости внеклеточного матрикса для IgG в SCIG добавляется рекомбинантная человеческая гиалуронидаза (rHuPH20), что позволяет сократить расход препарата на курс лечения пациента и добиться более высоких уровней антител в плазме крови [9]. По совокупности свойств и благодаря более простому применению в клинике, SCIG, особенно препараты с rHuPH20, способны вытеснить ВВИГ с рынка фармацевтических препаратов.
Существует серьезное противоречие между требованиями к качеству и эффективности ВВИГ, и его безопасностью. Прежде всего, оно имеет отношение к вирусной безопасности ВВИГ и других препаратов крови. Чтобы ВВИГ был эффективным и соответствовал критериям качества, плазма должна быть получена от как можно большего количества доноров, более 1000. Но чем больше донаций использовано для получения плазмы, тем больше риск того, что она будет инфицирована опасными для человека вирусами. Когда ВВИГ начали применять в клинической практике, считалось, что если IgG фракционируют холодным этанолом, то это обеспечивает вирусную безопасность полученных препаратов. Но эти надежды не оправдались, вирусы, особенно возбудитель гепатита С, продолжали находить в крупных партиях ВВИГ [19].
Таблица 2
Сравнение свойств SCIG и ВВИГ [14]
|
Свойство
|
ВВИГ
|
SCIG
|
|
Стабильность поддержания уровня IgG в сыворотке крови
|
Невозможно
|
Возможно
|
|
Возможность пиков концентраций IgG в сыворотке крови
|
Возможно
|
Невозможно
|
|
Защита от возбудителей инфекционных болезней
|
Возможна
|
Возможна
|
|
Доступ к венам
|
Нужен
|
Не нужен
|
|
Продолжительность инфузии
|
Несколько часов
|
Один час или меньше
|
|
Частота инфузий
|
Каждые 2–4 недели
|
Обычно одно введение в неделю
|
|
Возможность домашнего введения
|
Возможно, но мало кто из пациентов пользуется такой возможностью
|
Возможно, большинство пациентов предпочитает именно такое введение
|
Таблица 3
Способы, используемые для инактивации вирусов в препаратах крови [5, 12]
|
Способ
|
Суть способа
|
Недостатки способа
|
|
1
|
2
|
3
|
|
Пастеризация жидкого продукта
|
Нагревание продукта при температуре 60 °С в жидком состоянии 10–12 ч
|
Риск заражения вирусами гепатитов В и С существует при
использовании пастеризованных концентратов; необходимость использования
больших концентраций протекторов, в основном углеводов с различными
добавками, для защиты лабильных белков плазмы от денатурации.
Одновременно они стабилизируют и вирусы
|
|
Химическая инактивация вируса в жидком продукте
|
Основан на способности химических веществ проникать через липидную оболочку вирусов
|
Применение ограничено из-за лабильности белков плазмы крови.
Способ применим для инактивации вирусов, имеющих липидную оболочку.
Малоэффективен для инактивиции вирусов, вызывающих гепатит А или
парвовирусную инфекцию
|
|
Химическая обработка + ультрафильтрация продукта
|
Присоединение к химической обработке метода ультрафильтрации
|
Удаление продуктов распада вирусов требует дополнительных этапов
|
|
Ультрафиолет + химические вещества
|
Плазму и ее препараты подвергают облучению светом в
ультрафиолетовом диапазаоне в присутствии малых концентраций химических
веществ – красителей (метиленовый синий и др.)
|
Способ применим для инактивации вирусов, имеющих липидную
оболочку. Малоэффективен для инактивиции вирусов, вызывающих гепатит А
или парвовирусную инфекцию. Удаление продуктов распада вирусов требует
дополнительных этапов
|
|
Обработка лиофилизата паром
|
Лиофилизат подвергают обработке горячим паром в закрытой системе, заполненной инертным газом под давлением в течение 1–10 ч
|
В некоторых препаратах обнаруживали вирус гепатита В
|
|
Сухой прогрев лиофилизата
|
Инактивация вирусов происходит при нагреве лиофилизата при температуре 68 °С в течение 32–60 ч
|
В концентрированных негомогенных препаратах сохранялись вирусы гепатитов В, С и ВИЧ
|
|
Жесткая термообработка лиофилизата
|
Прогрев до 80 °С в течение 72 ч
|
Требуются протекторы, возможна частичная денатурация белков.
Возможно сохранение в препарате вирусов, вызывающих гепатит А или
парвовирусную инфекцию
|
Таблица 4
Показатели качества ВВИГ [12]
|
Показатель
|
Норма
|
|
Растворимость лиофильно-высушенного продукта
|
Менее 10 мин при температуре 20 °С
|
|
Прозрачность
|
Полная, не должно быть взвешенных частиц и невидимых частиц установленного размера
|
|
рН
|
4,0–4,5 или 6,8–7,4
|
|
Осмотическое давление, мОсм×моль/кг
|
≥ 240а
|
|
Общий белок, г/л
|
Не менее 30а
|
|
Содержание гамма-глобулина, %
|
≥ 95а
|
|
Димер + мономер, %
|
≥ 90а
|
|
Агрегаты, %
|
≤ 3а
|
|
Вспомогательные вещества (например, натрия хлорид, глицин, мальтоза, маннит, сахароза и т.п.)
|
В соответствии с нормативным документом
|
|
Вирус-инактивирующие агенты(сольвенты, детергены, три(n-бутил)фосфат, мкг/мл
|
≤ 10 или в соответствии с нормативным документом
|
|
Полисорбат 80, мкг/мл
|
≤ 100 или в соответствии с нормативным документом
|
|
Титр гемагглютинина Анти А
|
Негативный при разведении 1/64 (при концентрации белка 3 %)а
|
|
Титр гемагглютинина Анти В
|
Негативный при разведении 1/64 (при концентрации белка 3 %)а
|
|
Титр гемагглютинина Анти D
|
≤ 1а
|
|
IgА
|
В соответствии с нормативным документом. Максимальное количество должно быть указано на упаковке препаратаа
|
|
Активатор прекалликреина (prekallikrein activator, PKA), МЕ/мл
|
Менее 35 (при концентрации белка 3 %)а
|
|
Тест на идентичность человеческому происхождению
|
Положительныйа
|
|
Антикомплементарная активность, CH50⁄мг IgG
|
≤ 1а
|
|
Целостность Fc-фрагмента
|
Рутинного способа анализа не существует, используют применяемый при производстве иммуноглобулина
|
|
Титры специфических антител
|
В соответствии с регистрационным удостоверением лекарственного средстваб
|
|
HBsAg
|
Отсутствует а
|
|
Антитела к ВИЧ 1 и 2
|
Отсутствуют
|
|
Антитела к HCV
|
Отсутствуют
|
|
Бактериальная стерильность
|
Стерилен а
|
|
Тест на эндотоксин, МЕ/мл
|
Менее 0,5 (при содержании белка 5 %), менее 1,0 (содержание белка 5–10 %) а
|
|
Anti-HBs, МЕ/мл
|
≥ 0,5 а
|
|
Тест на пирогенность (LAL-тест)
|
Апирогенен
|
Примечания. а – European Pharmacopoeia specification; б – при
концентрации белка в препарате 50 г/л он должен содержать как минимум
два специфических антитела (одно к возбудителю вирусной инфекции, другое
– к бактериальной) в концентрации, по крайней мере, в три раза более
высокой, чем в стандартных препаратах ВВИГ.
Почти все мировые производители препаратов крови сталкивались с проблемой вирусной контаминации своих продуктов [5]. В настоящее время решение этой проблемы осуществляется в двух направлениях: адекватная проверка исходного сырья и применение все более совершенных технологий инактивации вирусов. Тем не менее проблема инфицирования больших пулов плазмы и, как следствие, получаемых из них препаратов, осталась. В табл. 3 приведены сведения об основных способах, используемых для инактивации вирусов в препаратах крови.
По мнению В.П. Панова [5], вирусная безопасность продуктов крови обеспечивается тремя непременными факторами: отбором доноров, тестированием стадии донации и пулов плазмы, инактивацией и удалением вирусов в процессе производства препаратов крови. Процедуры, обеспечивающие выполнение этих факторов, должны соответствовать правилам GMP. Максимальная степень вирусной безопасности может быть достигнута путем комбинации всех трех факторов. Обязательной является процедура валидации способов инактивации и удаления вирусов.
В то же время более чем 30-летний опыт разработки способов вирусной инактивации в препаратах крови показывает, что вряд ли удастся найти какой-то единый подход к инактивации оболочечных и безоболочечных вирусов. Современные технологические процессы производства препаратов из плазмы крови доноров должны включать, как минимум, две эффективных стадии вирусной редукции, причем одна из них должна быть эффективна против безо болочечных вирусов [4].
В целом же данные, приведенные в табл. 3, показывают, что до настоящего времени не разработаны технологические приемы, позволяющие гарантировать вирусную безопасность крови на 100 %. Любым их сочетанием достигается только «максимальная степень вирусной безопасности» (см. [5, 12]). Риск контаминирования вирусами препаратов крови возрастает при масштабировании производства, использовании концентрированных препаратов и расширении круга производителей.
Критерии качества, которые должен соблюдать производитель ВВИГ, определяет национальная фармакопея. Методы, используемые при оценке качества ВВИГ, должны быть валидированы. Показатели качества ВВИГ приведены в табл. 4.
Стандарты ВОЗ и референс-препараты ВВИГ и других препаратов крови европейские производители получают из Национального института биологических стандартов и контроля (National Institute for Biological Standards and Control, NIBSC; Potters Bar, UK), Центра оценки и исследований биологических препаратов Управления по санитарному надзору за качеством пищевых продуктов и медикаментов (Center for Biologics Evaluation and Research, Food and Drug Administration; CBER⁄FDA, Bethesda, MD) или из Европейского директората по качеству лекарственных средств и здравоохранения (European Directorate for the Quality of Medicines and HealthCare; EDQM, Strasbourg, France). Каталоги международных стандартов доступны на сайтах NIBSC и ВОЗ.
В то же время, современная концепция получения ВВИГ как пула IgG с высокой степенью очистки, выделенного из плазмы тысяч доноров (ВВИГ четвертого поколения), содержит ряд ограничений, неразрешимых в ее собственных рамках.
Сходство различных партий ВВИГ по физико-химическим свойствам, как это показано в табл. 4, не означает, что сходным будет и терапевтический эффект после их введения пациенту. Даже по приблизительным оценкам в стандартных поливалентных ВВИГ могут определяться антитела к 17 бактериальным антигенам, 21 антигену вирусов, 6 антигенам грибов и простейших [13]. Невозможно добиться воспроизводимых результатов, экспериментируя системой, в которой действуют по неизвестным закономерностям не менее 54 неизвестных факторов.
Иммунный ответ на введение ВВИГ трудно прогнозировать. Высокие дозы ВВИГ способны угнетать продукцию интерлейкинов (ИЛ) и снижать уровень экспрессии рецепторов к ИЛ2 вследствие наличия в препарате соответствующих антител, а также блокировать Fc-рецепторы при активных Fc-фрагментах IgG. Содержание в препаратах ВВИГ ряда биологически активных протеинов, например растворимых антигенов CD4, CD8 и антигена главного комплекса гистосовместимости (HLA) II класса, также оказывает влияние на реализацию физиологического иммунного ответа на антигенный раздражитель [3].
С позиций доказательной медицины в настоящее время подвергается сомнению терапевтическая эффективность ВВИГ при отдельной патологии. Например, ранее считалось, что при терапии сепсиса и септического шока можно использовать любые препараты, позволяющие нормализовать уровень иммуноглобулинов. Многолетняя клиническая практика и экспериментальные работы показали, что только введение ВВИГ, обогащенных IgM и IgA, повышает выживаемость пациентов. Однако IgA при случайном введении пациенту с врожденным дефицитом IgA, вызывает анафилактические реакции. В многоцентровом исследовании не обнаружено снижения летальности при использовании стандартных ВВИГ у кардиохирургических больных с тяжелой послеоперационной системной воспалительной реакцией, считавшейся показанием к их применению [12].
Существует проблема наличия в препаратах растворимых молекул CD4, CD8 и HLA II и I и антител к ним, которые могут влиять на распознавание антигена лимфоцитами; а также тромбина, факторов VIII, IX, X, XI и XII в количествах, активирующих свертывание крови и приводящих к образованию множественных тромбов у пациента [12].
Нерешенность этих проблем приводит к неопределенности результатов клинического применения ВВИГ четвертого поколения и требует их дальнейшего совершенствования, в том числе за счет расширения оцениваемых показателей качества. Одновременно можно предположить появление на рынке препаратов иммуноглобулинов иного типа: полученных генно-инженерным путем (без использования плазмы крови человека), поэтому хорошо охарактеризованных по молекулярному составу и обладающих высокой избирательностью в отношении мишеней воздействия.
Библиографическая ссылка
Супотницкий М.В., Елапов А.А., Борисевич И.В., Кудашева Э.Ю., Климов В.И., Лебединская Е.В. ИММУНОГЛОБУЛИНЫ ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ В АСПЕКТЕ ПОКАЗАТЕЛЕЙ КАЧЕСТВА, ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ // Успехи современного естествознания. 2015. № 5.
С. 84-94;
URL:
https://natural-sciences.ru/en/article/view?id=35106 (дата обращения: 11.07.2026).



