



В настоящее время в России вступила в действие фармакопея XIII, которая предусматривает селективное определение содержания тяжелых металлов в лекарственном растительном сырье (ЛРС) одним из современных инструментальных методов: атомно-абсорбционной спектрометрией, атомно-эмиссионной спектрометрией с индуктивно связанной плазмой (ИСП-АЭС) или масс-спектрометрией с индуктивно связанной плазмой [6]. Эти методы характеризуются общим этапом подготовки, предусматривающим перевод образца в раствор с помощью различных смесей на основе концентрированной азотной кислоты и других сильнодействующих реагентов. Выбор методики пробоподготовки во многом зависит от определяемого элемента и его концентрации, а также от природы органической матрицы ЛРС. Нами была разработана методика микроволновой минерализации цветков ромашки, которая характеризуется большей простотой и безопасностью по сравнению с методиками пробоподготовки, используемыми в элементном анализе [7]. По степени извлечения тяжелых металлов из растительного сырья в раствор она сопоставима с методикой ЕРА [10]. Цель данной работы – валидация разработанной методики пробоподготовки с целью ее применения в фармакопейном анализе при контроле качества цветков ромашки аптечной по показателю «содержание тяжелых металлов». Детектирование и количественное определение тяжелых металлов осуществляли методом ИСП-АЭС.
Материалы и методы исследования
Валидацию методики проводили на цветках ромашки аптечной, в которые предварительно (за 1 месяц до минерализации пробы) добавляли известные количества стандартных образцов (СО) определяемых элементов производства фирмы Merck (CRM) c аттестованным значением СО 1000 мг/дм3 (валидационные образцы). Для каждого уровня концентраций определяемых элементов готовили 3 параллельных пробы. Цветки ромашки без добавления СО тяжелых металлов использовали для приготовления холостого раствора. Минерализацию проб проводили концентрированной азотной кислотой аналитической степени чистоты (For Trace Metal Analysis), производства Acros Organics с помощью микроволновой системы Anton Paar Multiwave 3000. Содержание тяжелых металлов измеряли на атомно-эмиссионном спектрометре с индуктивно связанной плазмой Оptima 8300 DV фирмы Реrkin Elmer. Деионизованную воду, используемую при приготовлении испытуемых и стандартных растворов, получали на установке Milli-Q – Integral 3 фирмы Millipore, Франция.
Валидируемая методика
1,0 г (точная навеска) образца цветков ромашки, высушенных до постоянной массы при 105 °С в течение двух часов, растертых в однородный порошок и просеянных через сито с диаметром отверстий 1 мм, помещали в сосуд для микроволнового разложения и добавляли 8 см3 концентрированной азотной кислоты. При минерализации пробы использовали следующий временно-температурный режим: 0–80° (3 мин) → 80° (2 мин) → 80–165° (10 мин) → 165° (30 мин). Полученные после микроволнового разложения растворы охлаждали до комнатной температуры, фильтровали через фильтр «синяя лента» в мерные колбы объемом 25 см3 и доводили до метки деионизованной водой. Измерение содержания определяемых элементов проводили методом стандартных добавок, путем сравнения эмиссии испытуемого раствора и эмиссии растворов с добавками элементов известной концентрации. Характеристические длины волн эмиссии (λ) по выбранным элементам представлены в табл. 1. В случае As и Cd за результат принимали среднее значение концентраций, полученных по результатам двух длин волн. Статистические характеристики валидационных параметров рассчитывали, используя программное обеспечение MS Excel 2007.
Результаты исследования и их обсуждение
Валидацию методики пробоподготовки при количественном определении тяжелых металлов в цветках ромашки аптечной проводили согласно требованиям к валидации методик анализа [1, 5, 8]. В круг определяемых тяжелых металлов включили мышьяк, свинец, ртуть и кадмий, содержание которых в ЛРС нормируется многими национальными и международными нормативными документами [3]. Была проведена оценка линейности, предела количественного определения, правильности и прецизионности валидируемой методики. Специфичность и диапазон применения валидируемой методики пробоподготовки не оценивали, так как эти параметры лимитируются выбранным методом детектирования (для ИСП-АЭС они представлены в нормативной и научной литературе [2, 4, 9]).
Линейность
При определении линейности изучали зависимость измеряемой на атомно-эмиссионном спектрометре с индуктивно связанной плазмой интенсивности эмиссии элементов от содержания этих элементов в валидируемых образцах. Для оценки линейности проводили анализ серии из семи образцов цветков ромашки с прибавлением стандартных растворов тяжелых металлов с концентрациями 0,01; 0,02; 0,04; 0,08; 0,4; 0,8 и 1 мг/л (в рамках валидированной методики они соответствуют концентрациям 0,25; 0,5; 1; 2; 10; 20; 25 мг/кг элемента в образце). На основании полученных данных, представленных в табл. 1, были рассчитаны коэффициенты регрессионной прямой вида
y = bx + a,
где у – среднее значение измеренной на атомно-эмиссионном спектрометре с индуктивно связанной плазмой интенсивности эмиссии определяемых элементов; х – значение содержания элемента в валидируемом образце.
Графики регрессионных прямых и статистические характеристики установленных линейных регрессий приведены на рисунке.
Таблица 1
Результаты оценки линейности валидируемой методики
|
Концентрация СО, мг/л |
Средняя величина эмиссии |
|||
|
As (188,979 и 197,197 нм) |
Cd (214,440 и 226,502 нм) |
Pb (220,353 нм) |
Hg (194,168 нм) |
|
|
0,01 |
25,8 |
1847,7 |
108,1 |
– |
|
0,02 |
39,3 |
3734,0 |
215,6 |
119,0 |
|
0,04 |
81,0 |
7333,4 |
349,6 |
231,3 |
|
0,08 |
158,7 |
14500,1 |
586,5 |
467,3 |
|
0,4 |
811,4 |
75110,9 |
3023,7 |
2229,7 |
|
0,8 |
1603,4 |
149206,3 |
5948,7 |
4643,1 |
|
1,0 |
1907,9 |
180213,9 |
7127,9 |
6038,1 |
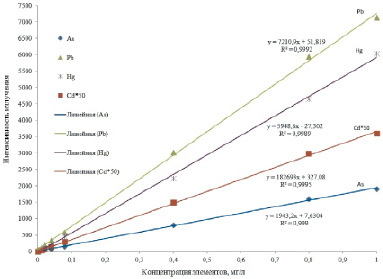
Линейная зависимость интенсивности излучения (эмиссии) элементов от их концентрации в образце
В соответствии с требованиями [5] критерием приемлемости линейной зависимости является коэффициент корреляции r ≥ 0,990. Из рисунка следует, что валидируемая методика характеризуется приемлемой линейностью по всем определяемым элементам.
Предел количественного определения (ПКО)
При проведении элементного анализа образцов ЛРС величины ПКО заметно возрастают из-за матричных эффектов, поэтому их необходимо оценивать в рамках конкретной методики. За ПКО принимали минимальную концентрацию элемента, для которой величина отношения сигнал/шум составила около 10:1 [1, 5, 8]. Значения ПКО As, Cd, Hg, Pb составляют 0,01; 0,001; 0,02; 0,01 мг/л соответственно.
Правильность
Для оценки правильности валидируемой методики использовали результаты, полученные в ходе установления линейности. Все данные были проверены на наличие выбросов по критерию Граббса. В соответствии с требованиями [1, 8] для всех образцов рассчитывали коэффициент извлечения – отношение «найдено: введено» Zi (табл. 2), стандартное отклонение, коэффициент вариации (или rsd), доверительный интервал и систематическую погрешность (табл. 3).
Правильность валидируемой методики оценивали по двум критериям приемлемости:
1) доверительный интервал должен включать 100 % значение коэффициента извлечения [1];
2) систематическая погрешность не должна превышать свой доверительный интервал (критерий статистической незначимости) [8].
Как видно из данных табл. 3, оба требования для анализируемых элементов выполняются, следовательно, валидируемая методика характеризуется приемлемой правильностью.
Прецизионность
Прецизионность оценивали на уровнях сходимости и внутрилабораторной прецизионности по результатам трех определений для каждого из трех уровней концентраций элементов. Полученные в условиях сходимости и внутрилабораторной прецизионности результаты измерения отношения «найдено:внесено» (Zi) и их статистической обработки представлены в табл. 4 и 5 соответственно.
Таблица 2
Результаты оценки правильности валидируемой методики
|
№ п/п |
Введено |
Определяемый элемент |
|||||||
|
As |
Cd |
Pb |
Hg |
||||||
|
Найдено |
Z, % |
Найдено |
Z, % |
Найдено |
Z, % |
Найдено |
Z, % |
||
|
1 |
0,01 |
0,007 |
70,0 |
0,009 |
90,0 |
0,012 |
120,0 |
– |
– |
|
0,013 |
130,0 |
0,010 |
100,0 |
0,011 |
110,0 |
– |
– |
||
|
0,009 |
90,0 |
0,009 |
90,0 |
0,011 |
110,0 |
– |
– |
||
|
2 |
0,02 |
0,024 |
120,0 |
0,020 |
100,0 |
0,024 |
120,0 |
0,018 |
90,0 |
|
0,020 |
100,0 |
0,021 |
105,0 |
0,022 |
110,0 |
0,016 |
80,0 |
||
|
0,020 |
100,0 |
0,020 |
100,0 |
0,023 |
115,0 |
0,017 |
85,0 |
||
|
3 |
0,04 |
0,037 |
92,5 |
0,041 |
102,5 |
0,046 |
115,0 |
0,040 |
100,0 |
|
0,042 |
105,0 |
0,040 |
100,0 |
0,047 |
117,5 |
0,037 |
92,5 |
||
|
0,044 |
110,0 |
0,041 |
102,5 |
0,044 |
110,0 |
0,035 |
87,5 |
||
|
4 |
0,08 |
0,088 |
110,0 |
0,078 |
97,5 |
0,081 |
101,3 |
0,078 |
97,5 |
|
0,084 |
105,0 |
0,080 |
100,0 |
0,085 |
106,3 |
0,079 |
98,8 |
||
|
0,082 |
103,0 |
0,081 |
101,3 |
0,073 |
91,3 |
0,079 |
98,8 |
||
|
5 |
0,4 |
0,370 |
92,5 |
0,386 |
96,5 |
0,377 |
94,3 |
0,396 |
99,0 |
|
0,354 |
88,5 |
0,399 |
99,8 |
0,375 |
93,8 |
0,378 |
94,5 |
||
|
0,373 |
93,3 |
0,380 |
95,0 |
0,386 |
96,5 |
0,388 |
97,0 |
||
|
6 |
0,8 |
0,705 |
88,1 |
0,729 |
101,1 |
0,727 |
90,8 |
0,815 |
101,9 |
|
0,734 |
91,8 |
0,790 |
98,8 |
0,793 |
99,1 |
0,816 |
102,0 |
||
|
0,745 |
93,1 |
0,778 |
97,2 |
0,776 |
97,0 |
0,799 |
99,9 |
||
|
7 |
1,0 |
0,886 |
88,6 |
0,921 |
92,1 |
0,895 |
89,5 |
1,027 |
102,7 |
|
0,897 |
89,7 |
0,938 |
103,8 |
0,942 |
94,2 |
1,041 |
104,1 |
||
|
0,899 |
89,9 |
0,941 |
94,1 |
0,942 |
94,2 |
1,065 |
106,5 |
||
Таблица 3
Статистические характеристики правильности валидируемой методики
|
Статистическая характеристика |
Определяемый элемент |
|||
|
As |
Cd |
Pb |
Hg |
|
|
Среднее значение Z, % |
97,7 |
98,4 |
103,6 |
96,5 |
|
Cистематическая погрешность δ = (Z – 100), % |
2,3 |
1,6 |
3,6 |
3,5 |
|
Cтандартное отклонение, % |
12,9 |
4,2 |
10,4 |
7,1 |
|
Коэффициент вариации, % |
13,2 |
4,3 |
10,0 |
7,3 |
|
Доверительный интервал (Р = 95 %), % |
97,7 ± 5,9 |
98,4 ± 1,9 |
103,6 ± 4,7 |
96,5 ± 3,5 |
Таблица 4
Результаты исследования сходимости и внутрилабораторной прецизионности методики
|
Внесено, мг/л |
0,02 |
0,4 |
1,0 |
||||||||
|
Параллельные измерения |
1 |
2 |
3 |
1 |
2 |
3 |
1 |
2 |
3 |
||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
Оператор 1 |
As |
найдено |
0,024 |
0,020 |
0,021 |
0,370 |
0,354 |
0,373 |
0,886 |
0,897 |
0,899 |
|
Zi, % |
120,0 |
100,0 |
105,0 |
92,5 |
88,5 |
93,3 |
88,6 |
89,7 |
89,9 |
||
|
Cd |
найдено |
0,020 |
0,021 |
0,020 |
0,386 |
0,399 |
0,380 |
0,921 |
0,938 |
0,941 |
|
|
Zi, % |
100,0 |
105,0 |
100,0 |
96,5 |
99,8 |
95,0 |
92,1 |
103,8 |
94,1 |
||
|
Pb |
найдено |
0,024 |
0,022 |
0,023 |
0,377 |
0,375 |
0,386 |
0,895 |
0,942 |
0,942 |
|
|
Zi, % |
120,0 |
110,0 |
115,0 |
94,3 |
93,8 |
96,5 |
89,5 |
94,2 |
94,2 |
||
|
Hg |
найдено |
0,020 |
0,016 |
0,017 |
0,396 |
0,378 |
0,388 |
1,027 |
1,041 |
1,095 |
|
|
Zi, % |
100,0 |
80,0 |
85,0 |
99,0 |
94,5 |
97,0 |
102,7 |
104,1 |
109,5 |
||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
|
Оператор 2 |
As |
найдено |
0,024 |
0,022 |
0,020 |
0,360 |
0,355 |
0,375 |
0,900 |
0,915 |
0,895 |
|
Zi, % |
120,0 |
110,0 |
100,0 |
90,0 |
88,8 |
93,8 |
90,0 |
91,5 |
89,5 |
||
|
Cd |
найдено |
0,020 |
0,020 |
0,020 |
0,380 |
0,396 |
0,395 |
0,920 |
0,941 |
0,935 |
|
|
Zi, % |
100,0 |
100,0 |
100,0 |
95,0 |
99,0 |
98,8 |
92,0 |
94,1 |
93,5 |
||
|
Pb |
найдено |
0,021 |
0,022 |
0,020 |
0,380 |
0,386 |
0,387 |
0,895 |
0,950 |
0,947 |
|
|
Zi, % |
105,0 |
110,0 |
100,0 |
95,0 |
96,5 |
96,8 |
89,5 |
95,0 |
94,7 |
||
|
Hg |
найдено |
0,020 |
0,017 |
0,017 |
0,391 |
0,386 |
0,375 |
1,021 |
1,020 |
1,044 |
|
|
Zi, % |
100,0 |
85,0 |
85,0 |
97,8 |
96,5 |
93,8 |
102,1 |
102,0 |
104,4 |
||
Таблица 5
Статистические характеристики сходимости и внутрилабораторной прецизионности валидируемой методики
|
Показатель |
Оператор 1 |
Оператор 2 |
||||||
|
As |
Cd |
Pb |
Hg |
As |
Cd |
Pb |
Hg |
|
|
Среднее Zi, % |
96,4 |
98,5 |
100,8 |
96,9 |
97,1 |
96,9 |
98,1 |
96,3 |
|
Стандартное отклонение, % |
10,5 |
4,4 |
11,1 |
9,3 |
11,0 |
3,2 |
6,1 |
7,2 |
|
Коэффициент вариации, % |
10,9 |
4,4 |
11,0 |
9,6 |
11,3 |
3,3 |
6,3 |
7,4 |
|
Доверит. интервал (Р = 95 %), % |
± 8,1 |
± 3,4 |
± 8,5 |
± 7,1 |
± 2,5 |
± 2,5 |
± 4,7 |
± 5,5 |
|
Объединенное среднее значение Zi, % As 96,8 Cd 97,7 Pb 99,5 Hg 96,6 |
||||||||
|
Объединенное стандартное отклонение, % * As 10,7 Cd 3,8 Pb 9,4 Hg 8,3 |
||||||||
|
Объединенный коэффициент вариации, % As 11,1 Cd 3,9 Pb 9,4 Hg 8,6 |
||||||||
|
Объединенный доверительный интервал, % As 7,6 Cd 2,7 Pb 7,4 Hg 5,9 |
||||||||
|
F-критерий Фишера |
As Fтабл = 3,44; Fфакт = 1,10 Cd Fтабл = 3,44; Fфакт = 1,83 Pb Fтабл = 3,44; Fфакт = 3,28 Hg Fтабл = 3,44; Fфакт = 1,69 |
|||||||
|
t-критерий Стьюдента |
As tтабл = 2,12; tфакт = 0,13 Cd tтабл = 2,12; tфакт = 0,85 Pb tтабл = 2,12; tфакт = 0,31 Hg tтабл = 2,12; tфакт = 0,15 |
|||||||
Примечание. *объединенные значения стандартного отклонения, коэффициента вариации и доверительного интервала с учетом данных двух операторов рассчитаны в соответствии с требованиями [8].
Статистические характеристики сходимости и внутрилабораторной прецизионности валидируемой методики представлены в табл. 5.
В случае оценки приемлемости внутрилабораторной прецизионности нормативные и методические документы в сфере GMP [1] рекомендуют рассчитывать статистические критерии Фишера (F) и Стьюдента (t) и сравнивать фактические значения tфакт и Fфакт с табличными – максимальными значениями критериев под влиянием случайных факторов при текущих степенях свободы и при заданном уровне значимости (tтабл и Fтабл). Как следует из данных табл. 5, табличные значения F и t превосходят фактические значения для всех определяемых элементов, что свидетельствует о статистической незначимости различий между средними значениями и стандартными отклонениями результатов измерений двух операторов при уровне значимости 95 %.
Таким образом, методика пробоподготовки при количественном определении тяжелых металлов в цветках ромашки аптечной методом ИСП-АЭС была оценена по основным валидационным параметрам, что доказывает возможность ее применения в фармакопейном анализе при контроле качества цветков ромашки аптечной по показателю «содержание тяжелых металлов».
Библиографическая ссылка
Щукин В.М., Северинова Е.Ю., Кузьмина Н.Е., Яшкир В.А., Меркулов В.А. ВАЛИДАЦИЯ МЕТОДИКИ ПРОБОПОДГОТОВКИ ПРИ КОЛИЧЕСТВЕННОМ ОПРЕДЕЛЕНИИ ТЯЖЕЛЫХ МЕТАЛЛОВ В ЦВЕТКАХ РОМАШКИ АПТЕЧНОЙ (MATRICĀRIA CHAMOMĪLLA) МЕТОДОМ ИСП-АЭС // Успехи современного естествознания. 2016. № 10. С. 57-62;URL: https://natural-sciences.ru/ru/article/view?id=36153 (дата обращения: 08.08.2026).